Cage escape governs photoredox reaction rates and quantum yields


Many photoredox studies use luminescence spectroscopy as a tool to monitor the reaction of an electronically excited catalyst with a substrate. Increasing substrate concentrations usually weaken the intensity of luminescence emitted by the catalyst, which is often interpreted as evidence of a successful photochemical reaction. However, luminescence quenching only monitors the disappearance of the excited state of the catalyst and does not contain direct information about the formation of photochemical products.
The “solvent cage”
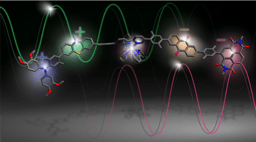
Photoinduced electron transfer between the catalyst and the substrate consumes energy and leads to quenching of the luminescence. The photoinduced electron transfer forms two short-lived radicals in close proximity to each other. These radical pairs are surrounded and held together by solvent molecules in a so-called “solvent cage” and tend to undergo spontaneous (light-independent) electron transfer in the reverse direction with each other. This undesirable reverse electron transfer restores the catalyst to its electronic ground state and the substrate to its original form. Light energy is absorbed and converted into heat, but no stable product is formed.
Radicals break free
To obtain stable products, the radicals formed in pairs must separate from one another by breaking free from the solvent cage. This elementary reaction step is called “cage escape” and its efficiency can vary greatly depending on the reaction. The slower the spontaneous reverse electron transfer within radical pairs, the more efficient the cage escape becomes1. In our case, this manifests in systematically higher cage escape quantum yields for a ruthenium(II) photocatalyst in comparison to a chromium(III) photocatalyst with many different reaction partners2. The driving-force associated with reverse electron transfer differs substantially between the two investigated photocatalysts, and this is likely the reason for the systematic differences in the cage escape quantum yields for ruthenium(II) and chromium(III)2,3.
More energy-efficient photochemistry
Recent work recognized the importance of cage escape for the formation of stable products in modern photoredox catalysis4,5. In our study, we find direct correlations between the quantum yields for cage escape and the efficiency of stable product formation. Three different photochemical reactions show substantially higher energy-efficiency with the ruthenium(II) photocatalyst compared to the chromium(III) photocatalyst, because of better cage escape with the ruthenium(II) photocatalyst. This fundamental insight opens new perspectives for more energy-efficient photochemical reaction design.
References
1. Gould, I. R.; Ege, D.; Moser, J. E.; Farid, S. Efficiencies of photoinduced electron-transfer reactions: role of the Marcus inverted region in return electron transfer within geminate radical-ion pairs. J. Am. Chem. Soc. 112, 4290-4301 (1990).
2. Wang, C.; Li, H.; Bürgin, T. H.; Wenger, O. S. Cage escape governs photoredox reaction rates and quantum yields, Nat. Chem. doi: 10.1038/s41557-024-01482-4 (2024).
3. Bürgin, T. H. ; Glaser, F.; Wenger, O. S. Shedding Light on the Oxidizing Properties of Spin-Flip Excited States in a CrIII Polypyridine Complex and Their Use in Photoredox Catalysis, J. Am. Chem. Soc. 144, 14181-14194 (2022).
4. Aydogan, A.; Bangle, R. E.; Cadranel, A.; Turlington, M. D.; Controy, D. T.; Cauët, E.; Singleton, M. E.; Meyer, G. J.; Sampaio; R. N.; Elias, B.; Troian-Gautier, L. Accessing Photoredox Transformations with an Iron(III) Photosensitizer and Green Light. J. Am. Chem. Soc. 143, 15661-15673 (2021).
5. DiLuzio, S.; Connell, T. U.; Mdluli, V.; Kowalewski, J. F.; Bernhard, S. Understanding Ir(III) Photocatalyst Structure-Activity Relationships: A Highly Parallelized Study of Light-Driven Metal Reduction Processes. J. Am. Chem. Soc. 144, 1431-1444 (2022).
Follow the Topic
-
Nature Chemistry

A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in