

Weaning is a critical stage in the life of a pig, particularly in current animal production systems that hit the animals with a double whammy of feed transition in combination with social stress caused by separation from the mother and littermates. Stress and the shift in the intestinal microbiota makes piglets susceptible to diarrheal pathogens, particularly Escherichia coli. Smoothing that transition will benefit well-being of piglets and reduce the need for antibiotics to treat enteric infections. Our publication provides a detailed analysis of the transition of swine microbiota after weaning – the combination of quantification of enzymes that relate to carbohydrate degradation with metagenomic binning allowed us to identify the key players in carbohydrate and fibre metabolism in the intestinal microbiome of swine.
We initially hypothesized that the use of probiotics beneficially influences the piglet’s microbiome, and that reutericyclin, an antimicrobial produced by Lactobacillus reuteri, enhances this impact. The impact of reutericyclin was assayed by the use of isogenic reutericyclin positive and –negative strains.
This proved to be yet another beautiful hypothesis that was slain by ugly facts.
Initial analyses demonstrated that the influence of probiotics on the overall gut microbiome was very minor; we had to look hard and with strain- or species specific to detect their influence on specific bacterial taxa. The reduction of pathogenic Escherichia coli that was observed in previous studies, however, could be confirmed.
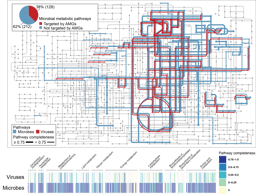
A more detailed analysis by metagenomic binning demonstrated that uncultured bacterial taxa represented most of the key organisms in the swine intestine – this indicates that much of the work that has been done for human intestinal microbiota, culturing of key bacteria to determine their physiological and metabolic properties – still remains to be done for swine intestinal communities. The “elephant in the room” explaining most of the changes in the microbiome was the diet – other factor were transient or did not matter. Consequently, our subsequent analyses focused on carbohydrate metabolism by gut microbiota. Because wheat and lactose were the major components of the feed, we focused on starch, fructans, and lactose as main dietary carbohydrates, and compared carbohydrate metabolism in the swine intestine to the corresponding organisms in the human intestine. The metagenomic assembly and binning that was performed by the collaborators from Huazhong Agricultural University provided an excellent and unprecedented foundation to analyse the metabolic networks for carbohydrate metabolism in the swine gut.
Comparable to human intestinal microbiota, starch degradation is dependent on metabolic cooperativity of diverse members of Firmicutes and Bacteroidetes. Surprisingly, the most abundant enzyme was not an amylase but a branching enzyme which likely functions to solubilise resistant starch and to make it accessible to hydrolytic enzymes.
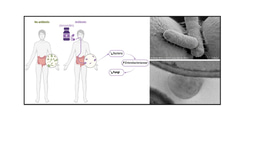
Key players for fructan and lactose metabolism in swine differed from those in the human intestinal microbiota. Lactobacilli expressing extracellular fructanases were major contributors to fructan metabolism in swine. Database searches revealed that the corresponding enzyme, FruA, is present in lactobacilli from the swine intestine but absent in all other lactobacilli – we have yet to figure out why this enzyme is important in pig intestine but not in any other of the (intestinal) ecosystems that harbor lactobacilli.
In the human gut and particularly in infants, bifidobacteria are the main degraders of lactose. In swine, lactobacilli take over that role and are major contributors to β-galactosidase activity. Taken together these findings may enable manipulation of the piglet’s microbiome after weaning by the animal’s diet.
The relationship between swine microbiota and lactobacilli in food fermentations provided an interesting and surprising sidekick of the work. Lactobacilli from swine are commonly found in specific cereal fermentations – in one of these fermentations, lactobacilli expressing the exceptional extracellular fructan hydrolase dominate and contribute to the degradation fructans for production of low-FODMAP bread. We were also surprised to find Lactobacillus delbrueckii at weaning. This organisms is used globally to produce yoghurt and has adapted to lactose and milk environments, however, to date, it has not been associated with intestinal environments. To confirm this unexpected occurrence, we went back to older datasets from weaning piglets; these analyses confirmed that L. delbrueckii is present at weaning but quickly disappears after the transition to solid feed. If confirmed by culture, this unexpected finding may demonstrate that L. delbrueckii adapted to suckling mammals, which could open new perspectives with regards to probiotics and intestinal health in infants and suckling animals.
I sometimes point out - tongue in cheek - that swine are an “in vivo model for food fermentations”. This study shows indeed that knowledge on host-adapted lactobacilli that is derived from food fermentations helps to explain metabolism in intestinal ecosystems. Conversely, knowledge on the ecology of these lactobacilli in the intestine helps us to improve their use in food fermentations and as probiotics.
Follow the Topic
-
Microbiome
This journal hopes to integrate researchers with common scientific objectives across a broad cross-section of sub-disciplines within microbial ecology. It covers studies of microbiomes colonizing humans, animals, plants or the environment, both built and natural or manipulated, as in agriculture.

Related Collections
With Collections, you can get published faster and increase your visibility.
Animal Gut Nutrition and Greenhouse Gas Mitigation
Animal Microbiome, Journal of Animal Science and Biotechnology and Microbiome call for submissions to the collection on Animal Gut Nutrition and Greenhouse Gas Mitigation.
Efforts to reduce greenhouse gas emissions from livestock systems increasingly hinge on innovations in animal gut nutrition. The dynamic relationship between the gut microbiome and nutrient utilization plays a pivotal role in shaping methane output, feed efficiency, and overall sustainability. Advances in microbial ecology—particularly in understanding the role of gut microbiome in nutrient metabolism—are opening new pathways for mitigating emissions while enhancing productivity. These developments support the implementation of climate-smart agricultural strategies to address climate change and its impacts.
Looking ahead, continued research in this field has the potential to yield innovative solutions such as targeted probiotic supplementation, which could further optimize gut function and enhance nutrient absorption. These advancements may lead to reduced greenhouse gas emissions while improving animal health and productivity. By deepening our understanding of the animal gut microbiome, we can contribute significantly to sustainable agricultural practices that benefit both the environment and food security.
We invite researchers to contribute to this special Collection on Animal Gut Nutrition and Greenhouse Gas Mitigation. Topics of interest include but are not limited to:
- Animal Gut Microbiome and Feed Efficiency
- Greenhouse Gas Mitigation Strategies
- Rumen Fermentation Dynamics
- Nutrient Utilization in Livestock
- Probiotic Supplementation Effects
- Sustainable Livestock Production Practices
- Climate-Smart Agriculture Innovations
This Collection supports and amplifies research related to SDG 13, Climate action.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Animal Microbiome fees, Journal of Animal Science and Biotechnology fees, Microbiome fees). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Publishing Model: Open Access
Deadline: Sep 04, 2026
Harnessing plant microbiomes to improve performance and mechanistic understanding
This is a Cross-Journal Collection with Microbiome, Environmental Microbiome, npj Science of Plants, and npj Biofilms and Microbiomes. Please click here to see the collection page for npj Science of Plants and npj Biofilms and Microbiomes.
Modern agriculture needs to sustainably increase crop productivity while preserving ecosystem health. As soil degradation, climate variability, and diminishing input efficiency continue to threaten agricultural outputs, there is a pressing need to enhance plant performance through ecologically-sound strategies. In this context, plant-associated microbiomes represent a powerful, yet underexploited, resource to improve plant vigor, nutrient acquisition, stress resilience, and overall productivity.
The plant microbiome—comprising bacteria, fungi, and other microorganisms inhabiting the rhizosphere, endosphere, and phyllosphere—plays a fundamental role in shaping plant physiology and development. Increasing evidence demonstrates that beneficial microbes mediate key processes such as nutrient solubilization and uptake, hormonal regulation, photosynthetic efficiency, and systemic resistance to (a)biotic stresses. However, to fully harness these capabilities, a mechanistic understanding of the molecular dialogues and functional traits underpinning plant-microbe interactions is essential.
Recent advances in multi-omics technologies, synthetic biology, and high-throughput functional screening have accelerated our ability to dissect these interactions at molecular, cellular, and system levels. Yet, significant challenges remain in translating these mechanistic insights into robust microbiome-based applications for agriculture. Core knowledge gaps include identifying microbial functions that are conserved across environments and hosts, understanding the signaling networks and metabolic exchanges between partners, and predicting microbiome assembly and stability under field conditions.
This Research Topic welcomes Original Research, Reviews, Perspectives, and Meta-analyses that delve into the functional and mechanistic basis of plant-microbiome interactions. We are particularly interested in contributions that integrate molecular microbiology, systems biology, plant physiology, and computational modeling to unravel the mechanisms by which microbial communities enhance plant performance and/or mechanisms employed by plant hosts to assemble beneficial microbiomes. Studies ranging from controlled experimental systems to applied field trials are encouraged, especially those aiming to bridge the gap between fundamental understanding and translational outcomes such as microbial consortia, engineered strains, or microbiome-informed management practices.
Ultimately, this collection aims to advance our ability to rationally design and apply microbiome-based strategies by deepening our mechanistic insight into how plants select beneficial microbiomes and in turn how microbes shape plant health and productivity.
This collection is open for submissions from all authors on the condition that the manuscript falls within both the scope of the collection and the journal it is submitted to.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Microbiome, Environmental Microbiome). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Collection policies for Microbiome and Environmental Microbiome:
Please refer to this page. Please only submit to one journal, but note authors have the option to transfer to another participating journal following the editors’ recommendation.
Collection policies for npj Science of Plants and npj Biofilms and Microbiomes:
Please refer to npj's Collection policies page for full details.
Publishing Model: Open Access
Deadline: Jun 01, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in