Beyond serology: unveiling the genetic blueprint of Escherichia coli’s outermost defense
Published in Microbiology, Protocols & Methods, and Genetics & Genomics
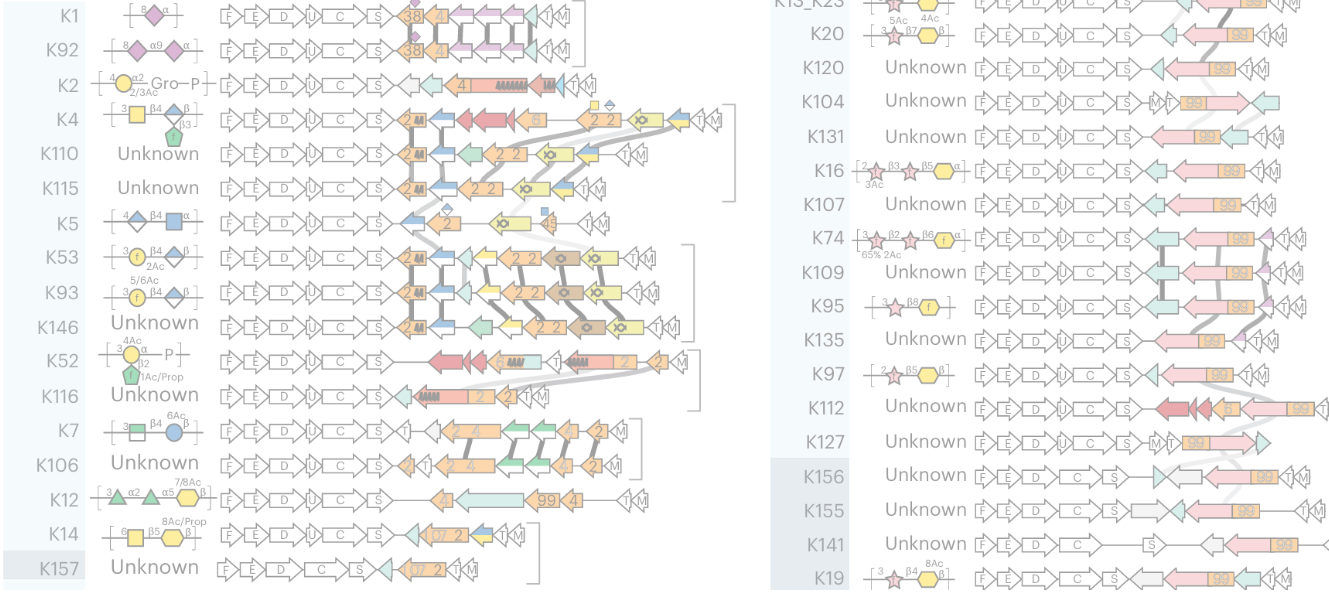
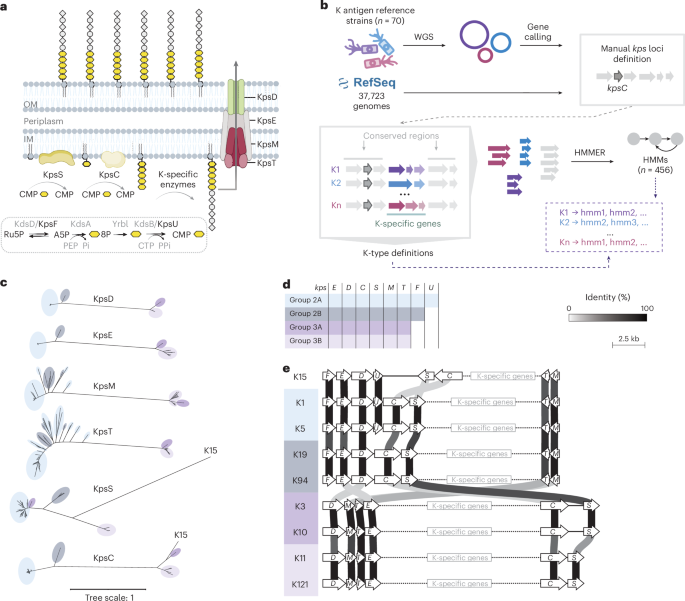
Capsular polysaccharides (K-antigens) shield Escherichia coli from the immune system. Despite their role in human disease, no new serotype has been described since 1977, and capsule epidemiology stagnated in the 1990s as scientists abandoned traditional serology. With this long-standing gap, a blind spot emerged around E. coli capsules. To bridge the gap, we first established a definitive genotype-serotype map for the 35 known transporter-dependent capsules. Then, we conducted an unprecedented genomic survey, analyzing over 37,000 E. coli genomes from diverse clinical and environmental sources, uncovering 55 additional novel K-antigens, including previously unrecognized evolutionary lineages. To map K-types at scale in genomes, we developed kTYPr, an in silico serotyping tool (Fig. 1). Leveraging Hidden Markov Models (HMMs), kTYPr accurately detects highly divergent or fragmented capsule gene clusters, outperforming other alignment algorithms and bioinformatic tools. kTYPr, deployed on a curated collection of > 26,000 genomes, uncovered a vastly greater diversity of E. coli K-types than previously recognized, particularly in understudied environments, revealing new associations with human disease.

kTYPr operates by scanning genome assemblies, whether isolated genomes or metagenome-assembled genomes (MAGs), against our curated reference database of known K-type loci and associated HMMs. The tool evaluates the presence, organization, and sequence match of key capsule-associated genes to assign the most likely K-type, while also flagging novel or divergent loci that do not match existing references. Its outputs include the predicted K-type per genome, confidence scores and match statistics, annotations of the capsule locus genes, and indicators of novelty. In large-scale analyses, kTYPr can also generate summary tables of K-type distributions across samples or environments, enabling downstream ecological and epidemiological comparisons (Fig. 2.).

Publicly available bacterial genomes reflects contexts where sequencing is needed and available: as a result, the bacterial diversity that we can study (for any trait, even beyond capsules) is heavily skewed towards bacteria isolated from humans rather than animal or environmental niches, from disease cases rather than healthy individuals, and from Western countries rather than low- and middle-income countries. We circumvented this problem by curating the metadata of a set of 32,043 E. coli genomes from NCBI aiming for individual host/niche resolution, and combining this information with genomic diversity (as 99% average nucleotide identity), to dereplicate the collection. This led to 23,188 entries, genomically and ecologically non-redundant, allowing us to explore capsule prevalence and associations with disease without artificially inflated numbers for heavily sampled niches or diseases.
The design of this collection revealed many previously unknown K-types in under-sampled niches such as food, livestock and wild animals, where up to half of detected capsule types had not been characterized before. This highlights the importance of extending sequencing efforts beyond heavily sampled niches such as human disease to discover new bacterial diversity. Conversely, the presence of known (and traditionally associated with disease) K-types in such undersampled niches underlines the importance of a One-Health perspective in the surveillance of bacterial pathogenicity traits, and pinpoints natural reservoirs that should be mapped for outbreak prevention. A key ecological insight is that capsule composition aligns more closely with ecological niche than with overall genomic relatedness. This suggests that capsule diversity is shaped by selective pressures such as host immunity, phages, and environmental conditions, combined with frequent horizontal gene transfer of capsule loci. Consistent with this view, ecologically generalist lineages exhibit particularly high diversity, reflecting ongoing adaptation across niches.
This newly discovered diversity has profound biomedical implications. For example, we uncover a complex relationship between capsule diversity and human health, showing that many capsule types traditionally associated with pathogenic strains (e.g. K1, K5) are also widespread in healthy gut microbiomes, which we could access via a globally distributed collection of 2,762 metagenome-assembled genomes from the stool of healthy individuals (Fig. 3A). This finding has been independently confirmed in distinct genome collections, in a parallel effort conducted by the Corander group (see https://www.nature.com/articles/s41564-026-02283-w). This suggests that strains harbouring pathogenic capsules can be successful commensals, “waiting” for an opportunity to invade. Beyond the old and new associations of K-types with invasiveness that we identified (Fig. 3B), we found many novel associations of other surface antigens, like O- and H-antigens, with disease. From these results, we would like to argue that capsule-associated pathogenicity is conditional and combinatorial, acting in concert with other bacterial and host factors. We hope that in the future bacterial pathogenicity for commensal-pathogens like E. coli will be evaluated with more matched carriage-invasiveness studies, to decouple host and bacterial determinants of invasiveness, ideally with time-resolved sampling of the host microbiome to “catch” the time and space where a commensal becomes a pathogen.

Fig. 3. K-type associations with human body niches and disease. A. Principal coordinates analysis (PCoA) of K-type profiles from different clinical categories, based on a Bray–Curtis dissimilarity matrix calculated from the proportion of K-types in each group. InPEC, intestinal pathogenic E. coli; MAGs, metagenome-assembled genomes; UTI, urinary tract infections; BSI, bloodstream infections. B. Odds ratio from a multivariable logistic regression model evaluating the association of K-types with invasiveness. The model adjusted for O-type, H-type, phylogroup, ST, host age, gender and geographic group, based on 4,923 E. coli genomes from distinct isolates and individuals including asymptomatic carriage (783 complete genomes and 2,762 MAGs) and invasive E. coli-associated disease (1,378 genomes from isolates from blood or cerebrospinal fluid).
We hope that kTYPr will help expand our knowledge of E. coli capsule epidemiology, to an extent comparable to the O- and H- antigens, that are routinely typed in clinical microbiology especially upon outbreaks. This will further test the new associations with invasiveness that we detected in our collections and provide an extended set of targets for capsule-targeting therapeutics, including drugs, phages, antibodies and vaccines for bacterial disease prevention, in an age where new antibacterial strategies are direly needed.
Check the full paper: https://www.nature.com/articles/s41564-026-02323-5
Download kTYPr: https://github.com/SushiLab/kTYPr
Follow the Topic
-
Nature Microbiology
An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in