Bioengineering innate immune cells
Published in Bioengineering & Biotechnology, Cancer, and Genetics & Genomics
A recently published paper on Nature Biotechnology about ‘Engineering Innate Immune cells for Cancer Immunotherapy” by Mubin Tarannum and colleagues drew my attention to the subject of cancer immunotherapy – yet again [Tarannum M. et al. 2025]. Having previously published on the theme here on my writing collection, on Nature Portfolio, a while ago (to satisfy a curiosity), and to highlight the clinical effects of immunomodulatory materials bioengineered in the lab - in November 2017. I followed up on the theme, with a more recent essay about the impact of epigenetic engineering for oncotherapy in June 2023. Once more out of curiosity, I am writing this article inspired by the same direction. I was further prompted to write the content since my current research direction as a basic and translational scientist/bioengineer is focused on designing biomedical research to investigate biomarkers of immunopathology in a bench-to-clinic translational context.
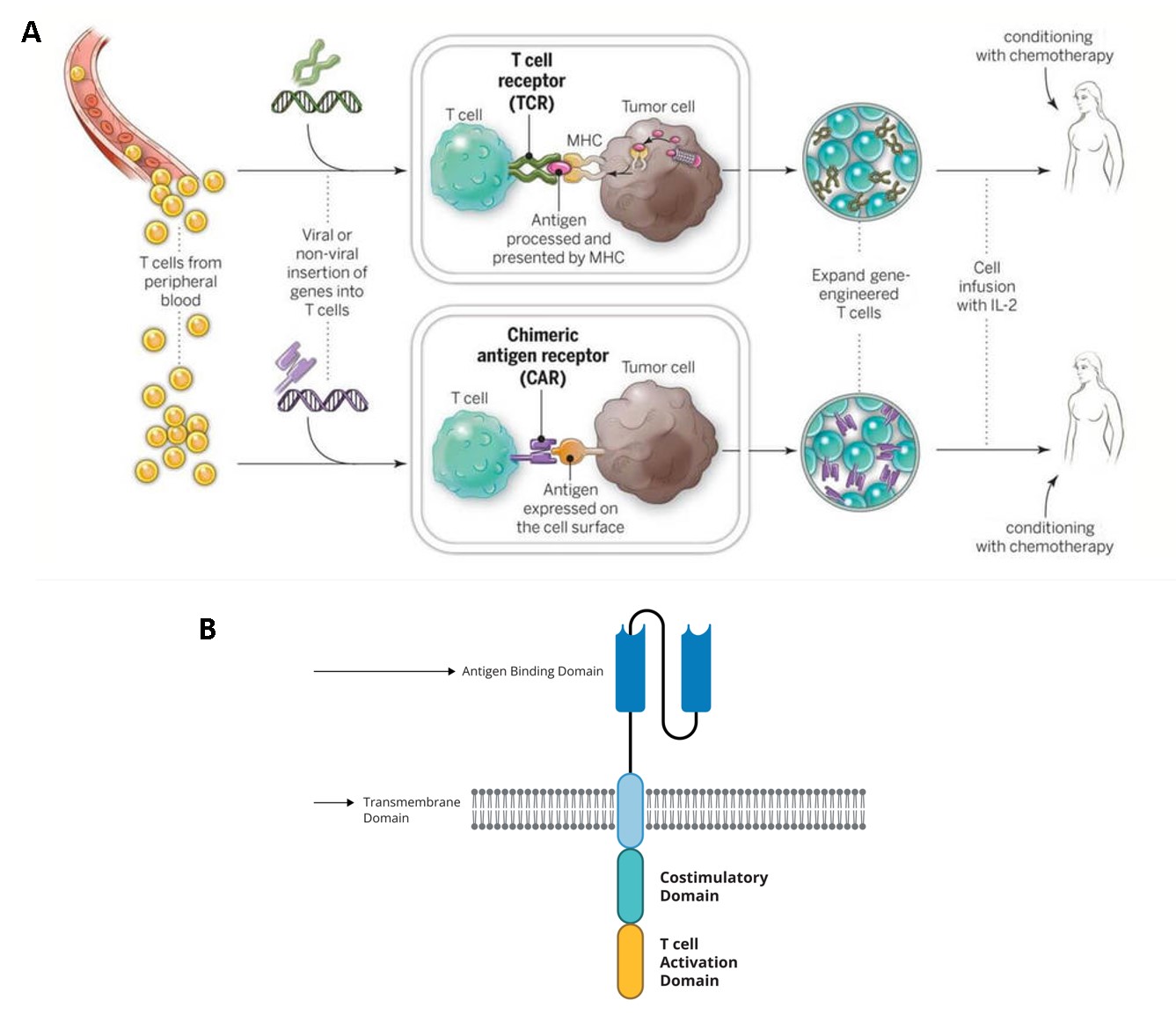
The more recent advances in the field of immuno-engineering have brought to light the capacity to repurpose innate immune cells, including natural killer cells, macrophages, and T cells as potential candidates for cancer immunotherapy [Tarannum M. et al. 2025]. In fact, chimeric antigen receptor T-cell therapy (CAR-T) is not a new idea; the concept of engineering an artificial or bioinspired T cell receptor that combines antibody-derived variable regions with T cell receptor derived constant regions - to identify cancer antigens, was introduced about three decades ago by the Japanese immunologist Yoshikazu Kurosawa in 1987. CARs were designed to engineer the patient’s own immune system to treat their cancer (Figure 1) [NCI 2025]. By 2013, the immunotherapy method transformed cancer treatment to become the “breakthrough of the year” [Mitra A. et al. 2023] and by 2018 it was further lauded as the “advance of the year” [Lindner S. et al. 2020]. The capacity to manipulate innate immune cells that forerun adaptive immunity, for immunotherapy, and understand its dual characteristics - is important to target pro-tumorigenic immune cells without losing anti-tumor immunity [Cambi A. and Sugimura R., 2023].
Chimeric antigen receptors (CARs) from bench-to-bedside
CAR-T immunotherapy forms a prelude to the now-prolific concept of engineering innate immune cell types, where T cells collected from patients form the backbone of the treatment regime to create a “living drug” [NCI 2025]. Simply put, the basis of these treatments is developed by collecting blood from the patient and separating out the T-cells to genetically engineer the cells to produce special proteins on their surfaces known as chimeric antigen receptors or CARs that are bioinspired by T cell receptors (Figure 1). These specifically engineered CARs help attach T cells or assist them to latch onto specific proteins or antigens present on cancer cells (and to a lesser extent present on normal cells) to accurately facilitate the killing of cancer cells. The revamped cells can be grown and expanded to obtain millions of the engineered immune cell types as an expanded product, which can then be returned to the patient as a single infusion; this whole process takes approximately 3-5 weeks to complete, with capacity to treat cancers and other virus-infected lymphocytes [Mitra A. et al. 2023, Perica K. et al. 2025].
After the infusion, T cells will continue to expand in the patient’s body, while targeting the elimination of cancer cells with cell-surface target antigens. CAR-T therapy is also administered in melanoma and triple-negative breast cancer (TNBC) mouse models with collagen anchored, for both systemic and local immunity/circulation in the experimental specimen [Momin M. et al. 2019]. At present, the strategy has advanced to include several FDA-approved CAR-T cell therapies targeting the treatment of multiple myeloma, follicular lymphoma, and B-cell acute lymphoblastic leukemia (B-cell ALL) (Table 1).
The bench-to-bedside timeline progression of CAR-T cell therapy across time emphasizes its clinical success (Figure 2), with instances where second-generation CAR-T cell therapy was used to treat advanced follicular lymphoma, by genetically engineering T cells to recognize CD19 receptors on malignant B cells [Kochenderfer J. et al. 2010]. The first clinically successful CAR-T cell therapy was administered in 2010 in an adult patient, to treat refractory chronic lymphatic leukemia (CLL). The first pediatric patient to receive the CD19-CAR T cell therapy to treat B-ALL leukemia developed side-effects such as severe cytokine release syndrome (CRS), necessitating the administration of tocilizumab and an anti-IL6 receptor antibody approved for rheumatoid arthritis [Mitra A. et al. 2023].
|
CAR T-Cell Therapy |
Approved Use(s) |
|
Abecma (ide-cel) |
Multiple myeloma |
|
Aucatzyl (obe-cel) |
B-cell ALL/B-ALL (acute lymphoblastic leukemia , adult) |
|
Breyanzi (liso-cel) |
|
|
Carvykti (cilta-cel) |
Multiple myeloma |
|
Kymriah (tisa-cel) |
|
|
Tecartus (brexu-cel) |
|
|
Yescarta (axi-cel) |
|
Table 1: FDA-Approved CAR T-cell Therapies. Credit: National Cancer Institute.
More recent news has also seen the successful administration of a low-dose methotrexate rheumatoid arthritis drug to improve anti-tumor activity, by reducing tumor metastasis, to enhance the effects of radiation therapy and immune checkpoint blockade therapy, in mice [Yang R. et al. 2025]. In general, CD19-CAR T cell therapy demonstrates a high response rate in pediatric patients and young adults with B-ALL (B-cell acute lymphoblastic leukemia), despite a majority encountering relapse, and requiring the administration of a second infusion of CAR-T cells or chemotherapy [Mitra A. et al. 2023]. Comparatively, CD22-targeted CAR T cell therapy has demonstrated higher efficacy and favorable outcomes in pediatric and young adults when compared to the CD19-CAR-T trials alone [Fry T. et al. 2018]. Furthermore, when coupled with stem cell transplantation, the long-term impact of CD19/CD22 CAR therapy in patients with b-ALL is efficient [Schultz L. et al. 2022, Shah N. et al. 2021].
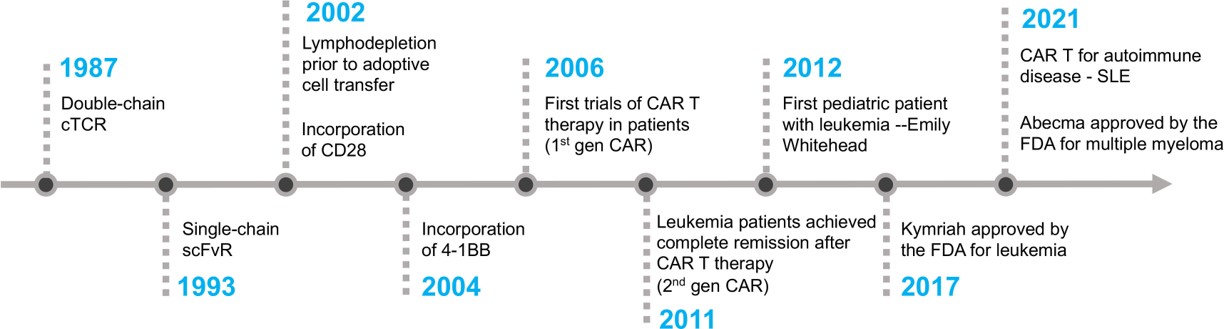
Figure 2: The timeline of key milestones accomplished during the development of CAR-T cell therapy [Mitra A. et al. 2023].
Four cutting-edge engineering approaches in the tumor microenvironment.
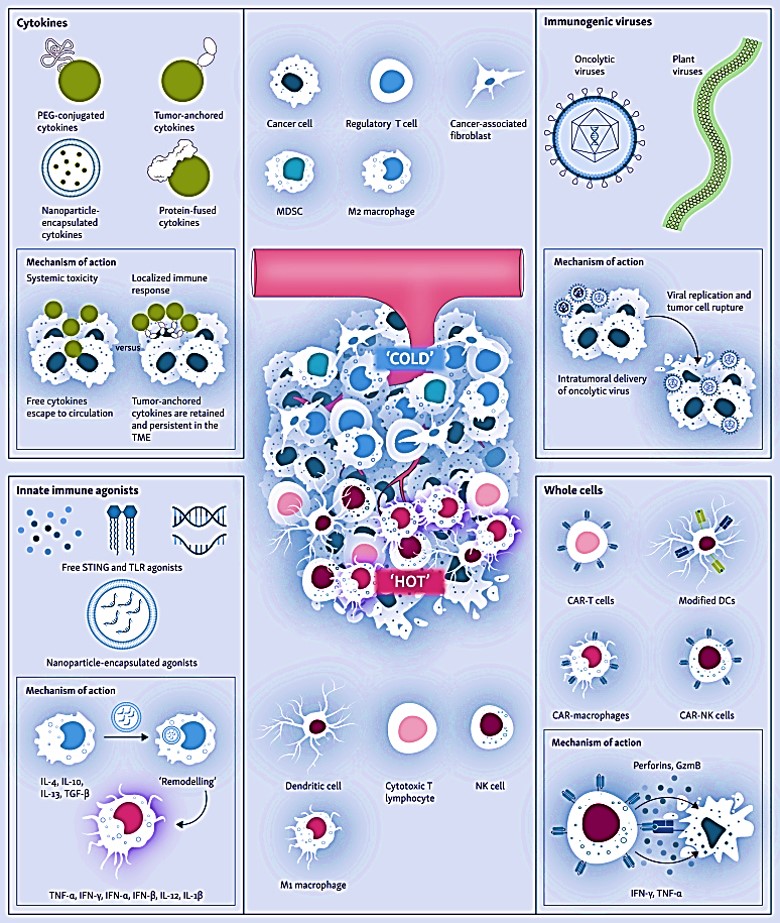
The capacity to engineer a correctly trained immune system through immunotherapy can offer transformative promise to treat lethal cancers by orchestrating tumor clearance through innate engineering, to ultimately transform the adaptive immune response; ideally without requiring continued therapeutic intervention. Four engineering approaches presently form the key focus of the immune-mediated tumor microenvironment remodeling process [Kane G. et al. 2023], to include:
- The production of cytokines such as interferons, transforming growth factor β (TGFβ) and tumor necrosis factor α (TNFα).
- A variety of chimeric antigen receptor (CAR) modified whole immune cells such as – T-cells (CAR-T), CAR-modified macrophages (CAR-M), and CAR-modified natural killer cells (CAR-NK).
- Innate immune agonists such as toll-like receptors (TLRs) and the stimulator of interferon genes (STING), as well as
- The integration of immunogenic viruses or oncolytic viruses for intra-tumoral delivery, and viral cell rupture (Figure 3).
The approval of the first CAR-T cell therapy, tisagenlecleucel (Kymriah) to treat relapsed or refractory acute lymphocytic leukemia, inadvertently set the tone for subsequent efforts in the niche of immune-engineering. While treatment strategies during early clinical trials showed the capacity to treat and eliminate leukemia in children, who were diagnosed with ALL [NCI 2025]. Long-term studies offered the outcome of a cure, enabling tisa-cel to be administered as a standard and recommended treatment for children who relapsed with ALL. The CAR-T therapies also received wide-spread attention and approval to treat blood cancers in adults, including multiple myelomas, and variations of lymphomas, based on large-scale clinical trials that emphasized the capacity to eliminate advanced cancers for a time in patients, while imparting cures in some (Table 1).
Nevertheless, these treatment methods are not without their limitations, where some can lead to severe side-effects including infections, and mass die-off of antibody producing B-cells that can be managed in most patients [NCI 2025]. As a result, a myriad of next-generation approaches has come to light – to both refine CAR T therapies and form chimeric antigen receptors (CARs) of other whole cells such as natural killer cells (CAR-NKs), macrophages (CAR-Ms), and develop dendritic cell vaccines (DC vaccines), to move at a brisker pace (Table 2).
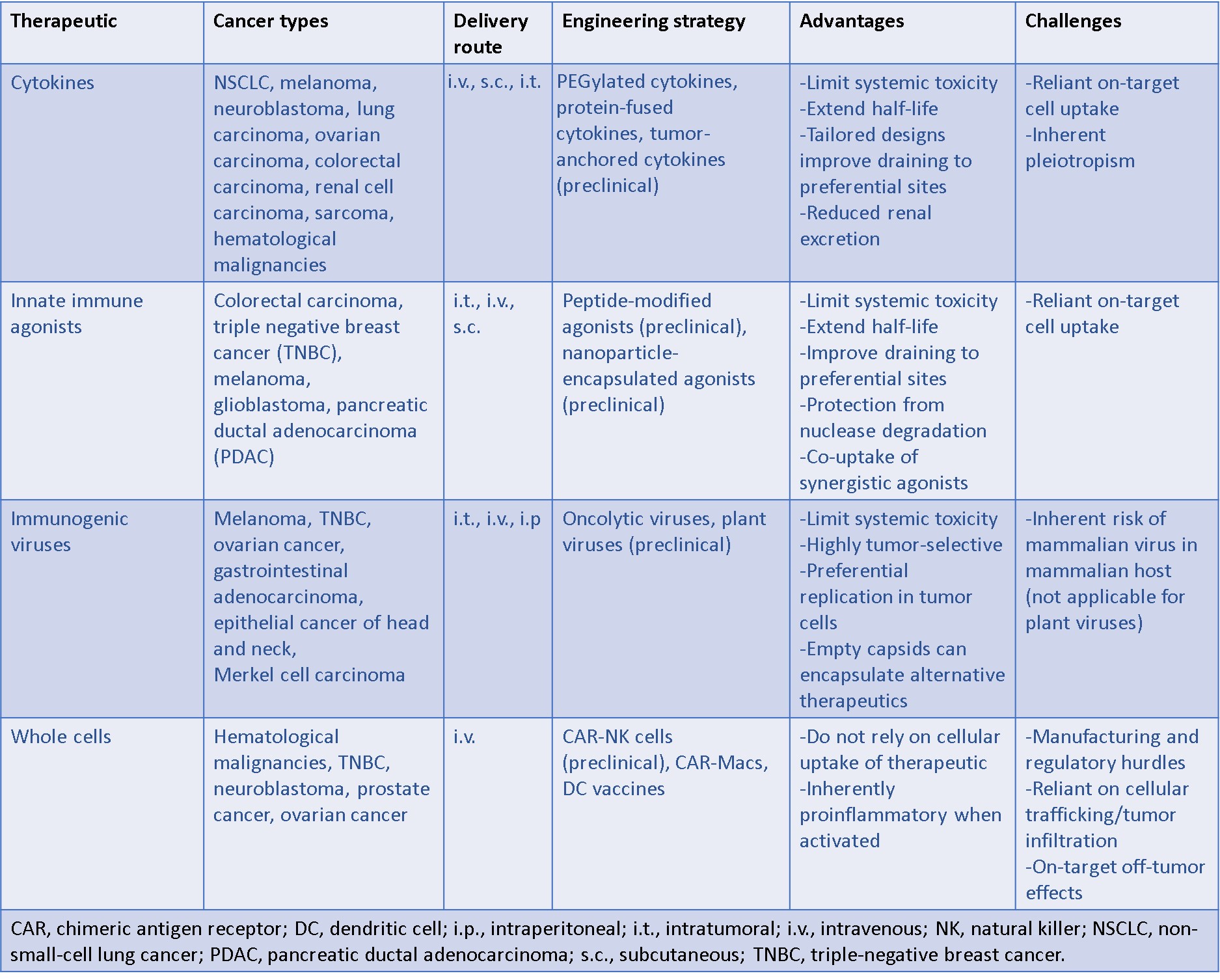
Whole-cell engineering– chimeric antigen receptor macrophages (CAR-M)
Large-scale cell-based approaches focus on the development of pro-inflammatory chimeric antigen receptors to bioengineer innate immune cells such as natural killer cells, and macrophages due to their significant potential for tumor microenvironment remodeling [Kane G. et al. 2023]. In addition to CAR T-based therapies, dendritic cells are also better suited as cell-based therapies since they can harness the professional antigen-presenting function during cancer vaccination [Wang Y. 2020]. Nevertheless, the technology must overcome significant manufacturing and regulatory challenges before their wide-spread applications in the clinic [Heathman T. et al. 2015]. These therapies are, however, limited since they rely on cellular trafficking and tumor infiltration, with CAR cells sometimes exhibiting off-target effects that can cause systemic inflammation [Sterner R. et al. 2021].
In a recent paper, therefore, Klichinsky and colleagues developed human chimeric antigen receptor macrophages (CAR-M) that can target the solid tumor antigens or anti-human epidermal growth factor receptor (HER2) to induce phagocytosis of the abnormal cancer cell types by producing M1-like macrophages (pro-inflammatory) for enhanced survival in an ovarian cancer model [Klichinsky M. et al. 2020]. During the experiments, the primary human macrophages were generated from peripheral blood CD14+ monocytes with granulocyte macrophage colony-stimulating factor (GM-CSF), to differentiate the M1-macrophage phenotypes in vitro. The anti-HER2 CAR was engineered into a vector to demonstrate vector-transduced human macrophages that expressed CAR with high efficiency and reproducibility between donors [Klichinsky M. et al. 2020]. During the study, the anti-human HER2 CAR-M macrophages showed antigen-specific phagocytosis of HER2+ beads and tumor cells. Thereby CAR-M therapy successfully eradicated an ovarian cancer cell line through a dose-and time-dependent manner, wherein the level of tumor phagocytosis correlated with the level of CAR-M expression (Figure 4).
By using a humanized tumor microenvironment (TME) mouse model, the effect of the engineered CAR-Ms can be interrogated at single-cell resolution. The work highlighted how the CAR-Ms induced several pro-inflammatory pathways, including interferon signaling in M2 macrophages to exert and maintain a dominant effect of anti-tumor activity in the surrounding immune cells. Since macrophages are professional antigen-presenting cells, bioengineers further tested the capacity of antigen-presentation with CAR-Ms to T-cells in vitro (Figure 5), with promising outcomes. The work additionally highlighted the potential to differentiate human peripheral blood monocytes (PBMCs) to macrophages endowed with tumor-antigen specific domains to traffic into tumors, to suggest the capacity to establish a CAR-M platform. In this precision therapeutic approach, CAR-Ms can directly diminish the tumor burden and sculpt the tumor microenvironment, while offering a vaccine-like effect [Klichinsky M. et al. 2020].
Engineering natural killer cells (CAR-NK) and dendritic cell vaccines
Unlike cytotoxic T-lymphocyte-associated proteins, natural killer cells are a more ideal immune cell type for chimeric antigen therapy due to their capacity to exert a powerful, pro-inflammatory cytokine response during tumor microenvironment remodeling, to ensure long-lasting anti-tumor immunity [Kane G. et al. 2023, Marofi F. et al. 2021]. For instance, yet again it is possible to treat the overexpression of epidermal growth factor receptor (EGFR) including the human epidermal growth factor receptor2 (HER2) associated with triple-negative breast cancers, by engineering natural killer cells as a specific chimeric antigen receptor [Liu Y. 2020]. As the cancers advance, triple-negative breast cancer cells (TNBC) can upregulate epidermal growth factor receptor signaling, for CAR-natural killer cells to then trigger the cell lysis of TNBCs in vitro and in mouse models - with similarities to the efficacy of CAR-M.
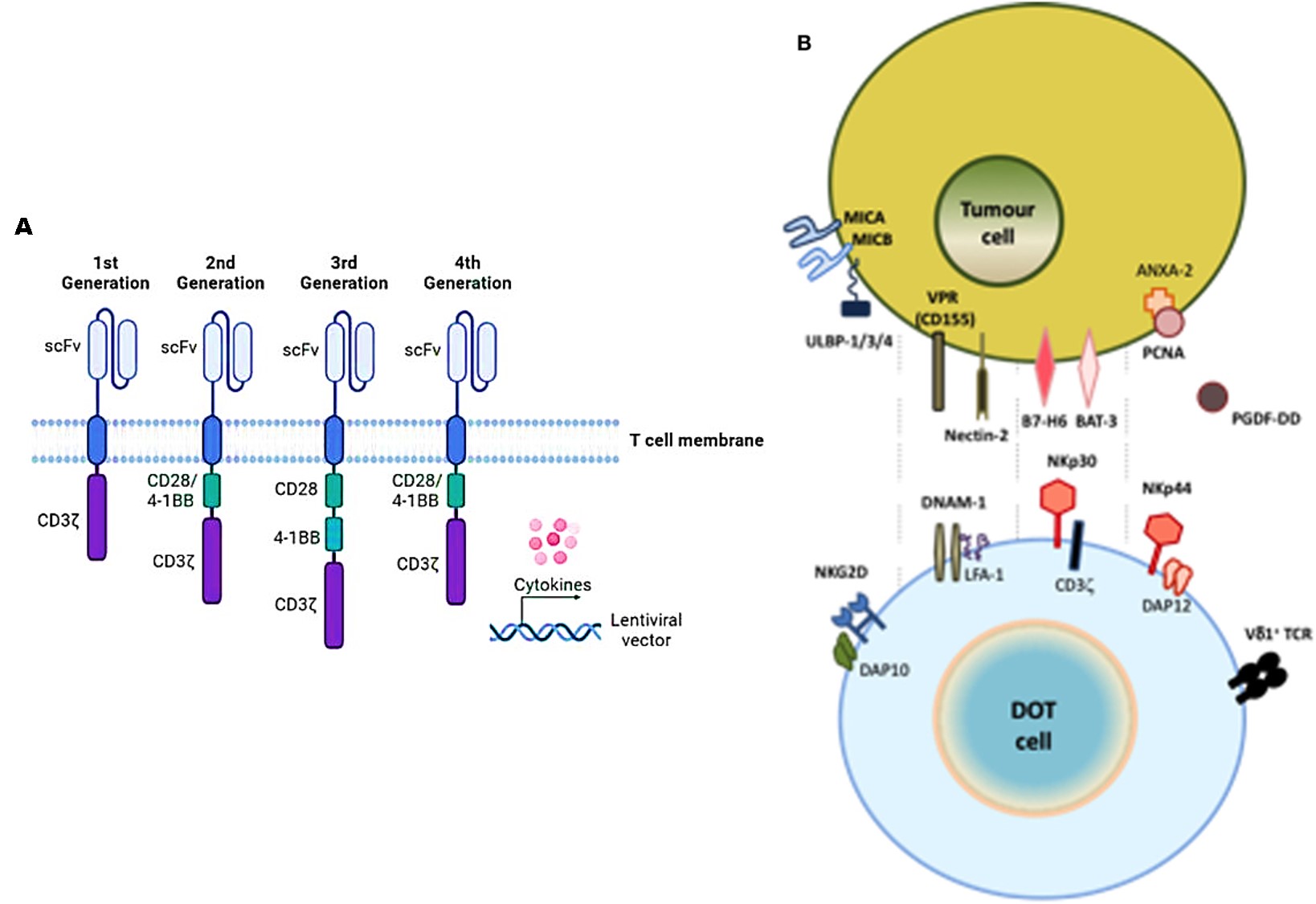
Efforts in bioengineering EGFR-CAR-NK cells in patient-derived xenograft mouse models of breast cancer, show a significant treatment effect as CAR-NK cells drive long-term tumor microenvironment (TME) remodeling [Liu Y. 2020]. More specifically, natural killer cells can also be engineered with a CAR receptor to activate the best characterized natural killer group 2 member D (NKG2D) receptor that can be fused to the cytotoxic ζ-chain of the T-cell receptor (TCR) to rescue impaired CAR-T cell activity against solid tumors (Figure 6). During its therapeutic implementation, both CAR-NK cells and CAR-T cells can be introduced consecutively to secrete pro-inflammatory cytokines in response to myeloid-derived suppressor cells (MDSCs) that are present in the solid tumor microenvironment - these cell types typically contribute to the suppression of cellular immunotherapy [Parihar R. et al. 2019]. This is followed by the infiltration and anti-tumor activity of CAR-T cells that are subsequently infused.
The possibility of combining the two strategies (CAR-NK, and CAR-T) affords a more promising mechanism for potential therapies. Proof-of-concept studies have shown how CAR-NK cells generated from pediatric neuroblastoma patients successfully eliminated the autologous MDSCs that typically suppress CAR-T cells in the tumor microenvironment [Feng S. et al. 2018]. Innovations that combine NK and T cells, show the possibility of establishing a pre-clinical proof-of-concept new cellular product known as delta one T cells (DOT) that are derived from γδ T cells for adoptive immunotherapy of cancer [Santos-Silva B. and Strid J. 2018]. DOT cells can upregulate NKG2D levels too, to induce the de novo expression of NK protein receptors that are associated with anti-tumor and anti-viral functions (Figure 6).
Dendritic cells are also crucial antigen presenting cells that play a significant role during immunotherapy by recognizing pathogenic antigens, to initiate immune responses [Wang Y. et al. 2020]. During anti-tumor responses, dendritic cell derived exosomes can participate in antigen presentation and exert an immune response, while facilitating crosstalk with natural killer cells. Dendritic cells are the most crucial sentinel cells for immune surveillance in the tumor microenvironment. For instance, a personalized dendritic cell vaccine can induce T cell immunity, to target tumor-specific regions, as a promising platform for precision medicine during tumor immunotherapy in patients with melanoma [Wang Y. et al. 2020]. In its mechanism-of-action, dendritic cells mediate tumor immunity by activating CD8+ and CD4+ T cells for tumor immunotherapy in preclinical and clinical studies to treat pancreatic cancer, glioblastoma, and HERpositive breast cancer to name a few examples (Figure 7) [Reap A. et al. 2018, Mehrotra S. et al. 2017, Lowenfeld L. et al. 2017].
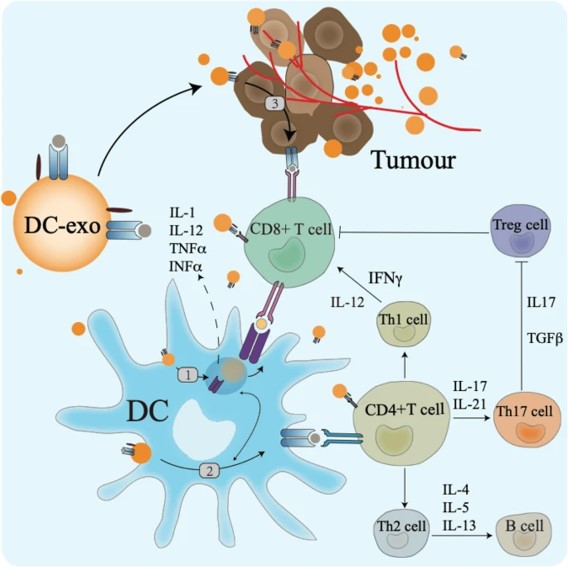
Curative tumor regression of large tumors is mainly mediated by CD8+ T cells and cross-presenting dendritic cells, to effectively engage the innate and adaptive immune responses to target abnormal cells in animal models [Moynihan K. et al. 2016]. Dendritic cell vaccines were tested during multiple clinical trials to target a variety of tumor-specific or tumor-associated antigens, including cytomegalovirus, telomerase, HER2 and Wilm’s tumor, to name a few [Wang Y. et al. 2020]. For example, researchers and clinicians have already implemented two stage I clinical pilot trials using vaccination with cytomegalovirus pp65-mRNA loaded dendritic cells in patients with glioblastoma [Reap A. et al. 2018]. Patients who received the dendritic cell vaccination experienced increased levels of interferon gamma, tumor necrosis factor alpha, and increased levels of CMV-specific CD8+ T cells to ensure long-term, progression-free survival, and overall survival rates [Batich K. et al. 2017].
The versatility of CAR-macrophages during immunotherapy across several diseases
Macrophages are a versatile and innate immune-mediated cell type, and I have broadly written several notes relative to my perspectives of their dual pathological and immuno-stimulatory roles during renal calcification in the kidney tissue, with capacity to hone-in their therapeutic value for clinical translation [Jeewandara T. 2025]. Macrophages are essentially sentinels and regulators of the immune system that can translate to cell-based therapies [Franken L. et al. 2016]. The inherent capacity of the cells to function as central regulators or effectors of innate immunity is accentuated by their role during phagocytosis, pro-inflammatory cytokine secretion, antigen presentation, and cytotoxicity, which can translate to precision molecular medicine platforms [Franken L. et al. 2016].
Having previously validated the experimental potential of differentiating the cell types in-lab, starting from peripheral blood mononuclear cells (PBMCs) into proinflammatory M1 and anti-inflammatory M2 macrophages in the presence of cytokines and colony-stimulating factors [Jeewandara T. 2025]. It is important to note several studies that emphasize the scope of regulating the classical M1 pro-inflammatory to the nonclassical M2 anti-inflammatory polarization, to attenuate disease phenotypes in lab. Examples include the M2-driven phagocytosis of calcium oxalate crystals in renal stone-forming patients - for their subsequent therapeutic applications in the clinic (Figure 8) [Taguchi K. et al. 2021].
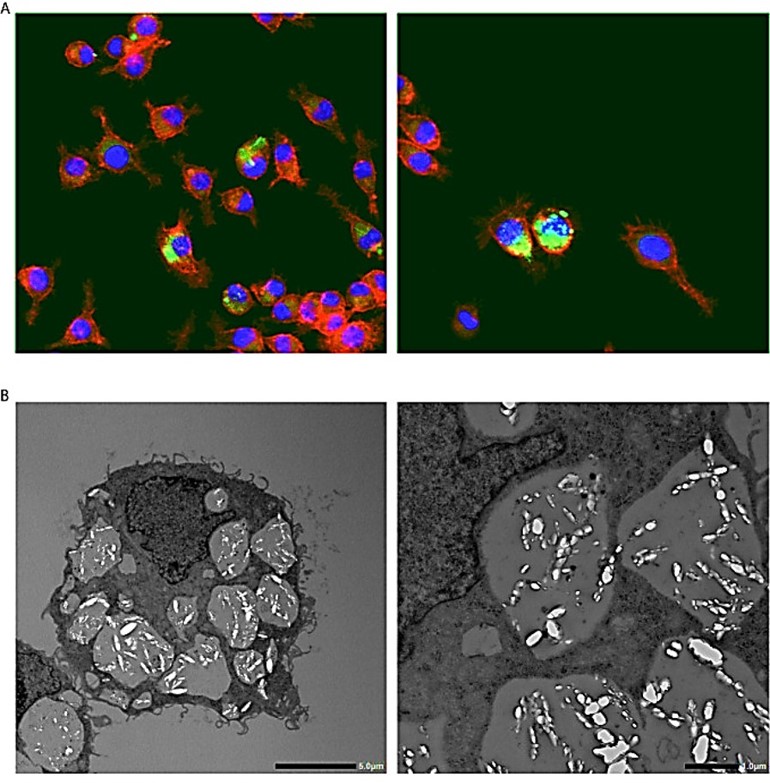
Figure 8: Calcium oxalate crystal phagocytosis by macrophages. A) Fluorescent immunohistochemical staining of macrophages cultured with calcium oxalate monohydrate crystals. Red – phalloidin, Green – calcium oxalate monohydrate crystals, Blue – nucleus. Magnification 400 X. B) Transmission electron microscopy images of macrophages that engulf calcium oxalate monohydrate crystals [Taguchi K. et al. 2021].
Conversely, during cancer immunotherapy, the opposite is true, where the M2 anti-inflammatory macrophages in the tumor-microenvironment are geared to polarize to the immunostimulatory/pro-inflammatory M1 phenotype, to allow the tumor-associated macrophages to be re-engineered as M1 immunotherapeutic adjuvants [Weiskopf K. et al. 2013]. However, these experimental efforts must consider that tumor-associated macrophages express both activating and inhibitory Fc receptors during cancer immunotherapy [Weiskopf K. et al. 2017]. The chimeric antigen receptor (CAR) macrophages (CAR-m) are unique due to their function as professional antigen presenting cells (APCs) that promote adaptive anti-tumor immune responses. Furthermore, CAR-macrophages accurately incorporate the M1 pro-inflammatory phenotype, as a driver of tumor microenvironment remodeling [Kane G. et al. 2023].
Dwelling deeper - genetically engineering human macrophages
Human macrophages can be genetically engineered with chimeric antigen receptors to direct their phagocytic function to attenuate several disease phenotypes and enable tumor microenvironment remodeling (Movie 1) [Klichinsky M. et al. 2020]. Since CAR macrophages uniquely bear the M1 pro-inflammatory phenotype by definition, to drive tumor microenvironment remodeling, they can circumvent tumor macrophage polarization by steadily switching macrophage phenotypes from M2 (anti-inflammatory) to the M1 pro-inflammatory phenotypes in vivo [Noy R. and Pollard J. 2014]. In its mechanism of action, CAR-M macrophages exert an adaptive immune response via an active process that requires spleen tyrosine kinase (SYK) – a non-muscle myosin II a, and actin polymerization process, much like the process of Fc receptor-mediated antibody dependent cell phagocytosis. As a result, it is possible to generate CAR-Ms that target solid tumor antigens mesothelin or HER2, to demonstrate phagocytosis of antigen-positive target cells, in solid tumors [Klichinsky M. et al. 2020, Hassan R. and Ho M., 2008].
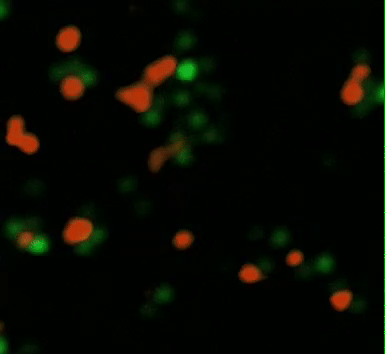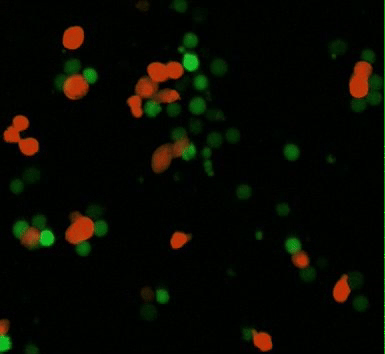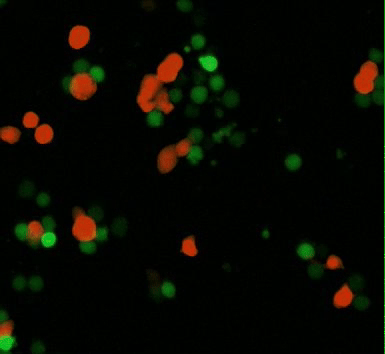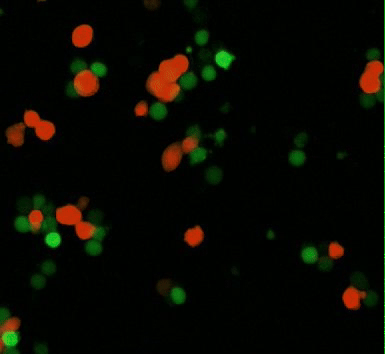
Movie 1: CAR-mediated macrophage phagocytosis, where CAR-Ms as a therapeutic platform show capacity of anti-tumor phagocytosis activity. Phagocytosis of a leukemia cell line with CAR-macrophages [Klichinsky M. et al. 2020].
The process of gene transfer into human macrophages as a method of CAR delivery is a long-standing challenge [Fleetwood A. et al. 2007]. Efforts to facilitate this process include using the expression of the cluster differentiation 46 (CD46) membrane cofactor protein, to functionalize protein molecules as a docking protein for group B adenoviruses, such as Ad35 to engineer a replication-incompetent chimeric adenoviral vector (Ad5f35), to deliver CAR to macrophages [Nilsson M. et al. 2004]. Recent studies have further demonstrated how anti-HER2 CAR could be engineered into an adenoviral vector (Ad5f35) to test its principle-of-action, to demonstrate that vector-transduced human macrophages expressed CAR efficiently. The primary anti-HER2-CAR-Macrophages can effectively phagocytose HER2+ tumor cells and eradicate a HER2+ ovarian cancer cell line (SKOV3), in a dose- and time-dependent manner. Again, the level of tumor phagocytosis correlates with the level of CAR expression. Meanwhile, on-target/off-tumor toxicity assays showed how normal cells were not phagocytosed by the anti-HER2-CAR-Macrophages, in mice [Klichinsky M. et al. 2020].
Cutting-edge developments in chimeric antigen receptor engineering
Many of the early approaches to engineering innate immune cells for targeted immunotherapy have presently reached a zenith to facilitate a range of treatment options via clinical oncotherapy. Today, the engineered CAR-T immune cells are a powerful therapy that oncologists incorporate to treat many types of blood cancers, with valuations of the technology expected to reach nearly $190 billion by 2034 [Willyard C. 2024]. Nevertheless, the CAR-T therapies come with a serious downside due to their labor-intensive development process and administration. For example, the efficacy of chimeric antigen receptor T cell (CAR-T) therapy against solid tumors is limited due to its immunosuppressive microenvironment. In contrast, CAR-macrophages can move efficiently to facilitate immunotherapy for solid tumors, although CAR-M is still at its nascent stages of clinical trials, with results already showing potential advantages of CAR-macrophage therapy vs. CAR-T, to treat solid tumors. In fact, this phenomenon is compared to a race between a rising star (CAR-M) and a superstar (CAR-T) [Chen K. et al. 2024].
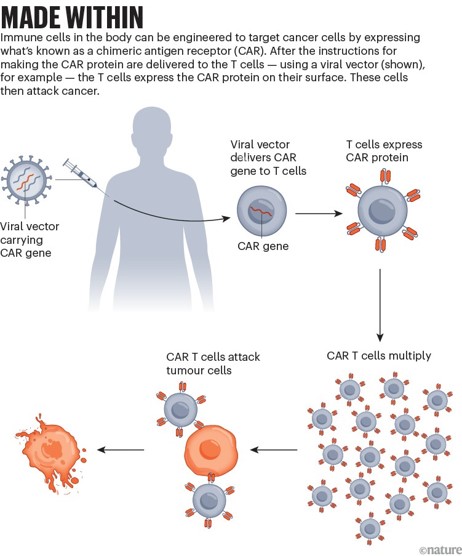
Made within
Investigating the relationships and differences among various immunotherapies can facilitate new insights to optimize the strategies of treatment. Newer methods to optimize CAR-T therapy, for instance, have introduced highly inventive measures such as altering T cells within the body itself, without having to extract them from people undergoing treatment (Figure 9) [Ledford H. 2023]. Engineering cells inside the body is, however, an intricate science. The in vivo CAR-T therapies are based on the ex-vivo playbook, where an engineered lentivirus grabs onto T cells to deliver the gene for the CAR protein into the genome of the cell, to be ‘made within.’ Targeting the cellular genome is cumbersome since there are a variety of common receptors in vivo, and bioengineers must specifically target T cells to get to the precise location at the accurate time. To accomplish this, one approach includes a vector that can latch onto the CD7 found on T cells, and on natural killer cells.
Another incorporates a lentiviral vector decorated with a protein that can target three receptors on T cells at once, to simplify the CAR-T therapy approach. By engineering cells in vivo, it is possible to overcome chemotherapy treatment, thereby eliminating chemotherapy-associated side-effects such as risks of infection [Willyard C. 2024]. Instead of viral vectors, it is also possible to ferry RNA into T cells via nanoparticles where the RNA enters the cell’s cytoplasm as genetic information, to build the CAR protein for a short timeframe for safer approaches in the form of multiple doses of a therapy. Such efforts can be commercialized to deliver RNA of the CAR proteins to T cells, or to natural killer cells and macrophages, to treat blood cancers. The CAR-T therapy can also be made faster and cost-effective (as seen in a home-grown biotechnology company in India), to produce CAR-T cells within 22 hours, for a simpler and more accessible process [Mallapaty S. 2024].
Sonogenetics and next-generation CARs at the molecular level
To wrap things up, it is worthwhile to highlight how CAR-T cells are being engineered with sonogenetics or ultrasound, to produce "EchoBack-CAR T cells" that can eliminate solid tumors with minimal toxicity via the use of focused ultrasound simulation [Liu L. et al. 2025]. Focused ultrasound is a highly effective and non-invasive therapeutic method in bioengineering that can transmit and amplify ultrasound sensitivity in target cells by integrating engineered or modified protein components. The capacity of ultrasound waves to pass through the body and deliver energy causes a wide range of bioeffects of low or high pressure, frequency, and duration, suited for a variety of applications including neuromodulation or targeted cancer immunotherapy. The method when guided by magnetic resonance imaging can precisely direct an ultrasound beam to an accurate location with high resolution, to target small volumes of cells in deep tissue environments such as tumor microenvironments to ablate cancerous tissue, facilitate drug delivery mechanisms or modulate the activity of excitatory neurons for precision medicine [Bell K. et al. 2023].
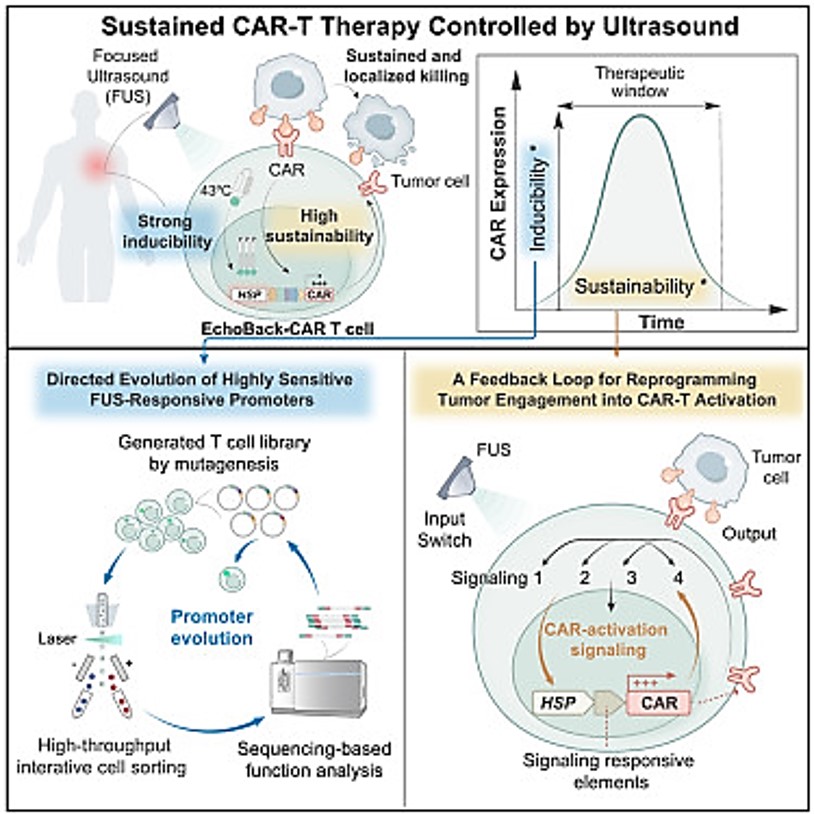
As highlighted in this article, although the treatment of blood cancers was clinically successful, CAR-based immunotherapy has faced significant challenges during the treatment of solid tumors. More specifically, the method must be able to target life-threatening non-specific CAR T cells against normal vs. malignant tissues. Recent advances in synthetic biology and genetics regulation have led to advances in sonogenetics to facilitate a precise activation of genes remotely, to mitigate off-target toxicity that is typically observed with CAR-T cell therapies to offer a safer and more effective platform to treat solid tumors [Liu L. et al. 2025].
In a similar method, the genetic transfer of CAR mRNA can be achieved through many approaches, including ultrasound activation, hypoxia, temperature and light regulation, small molecules as well as logic (AND)-gate mechanisms of synthetic biology (Figure 11) [Celichowski P. et al. 2023]. Early CARs contained antibody single chain variable fragments (scFvs) fused through a transmembrane domain to the cytoplasmic tail of the TCR signaling component CD3ζ signaling domain, which includes both Kymriah and Yescarta drugs as well as a variety of clinically used second generation CARs (Figure 6) [Lindner S. et al. 2018, Yun K. et al. 2023]. Each additional next-generation CAR constructs add further signaling capacity to the engineered receptors for greater efficacy, for off-the-shelf products (Figure 11) [Labanieh L. et al. 2018].
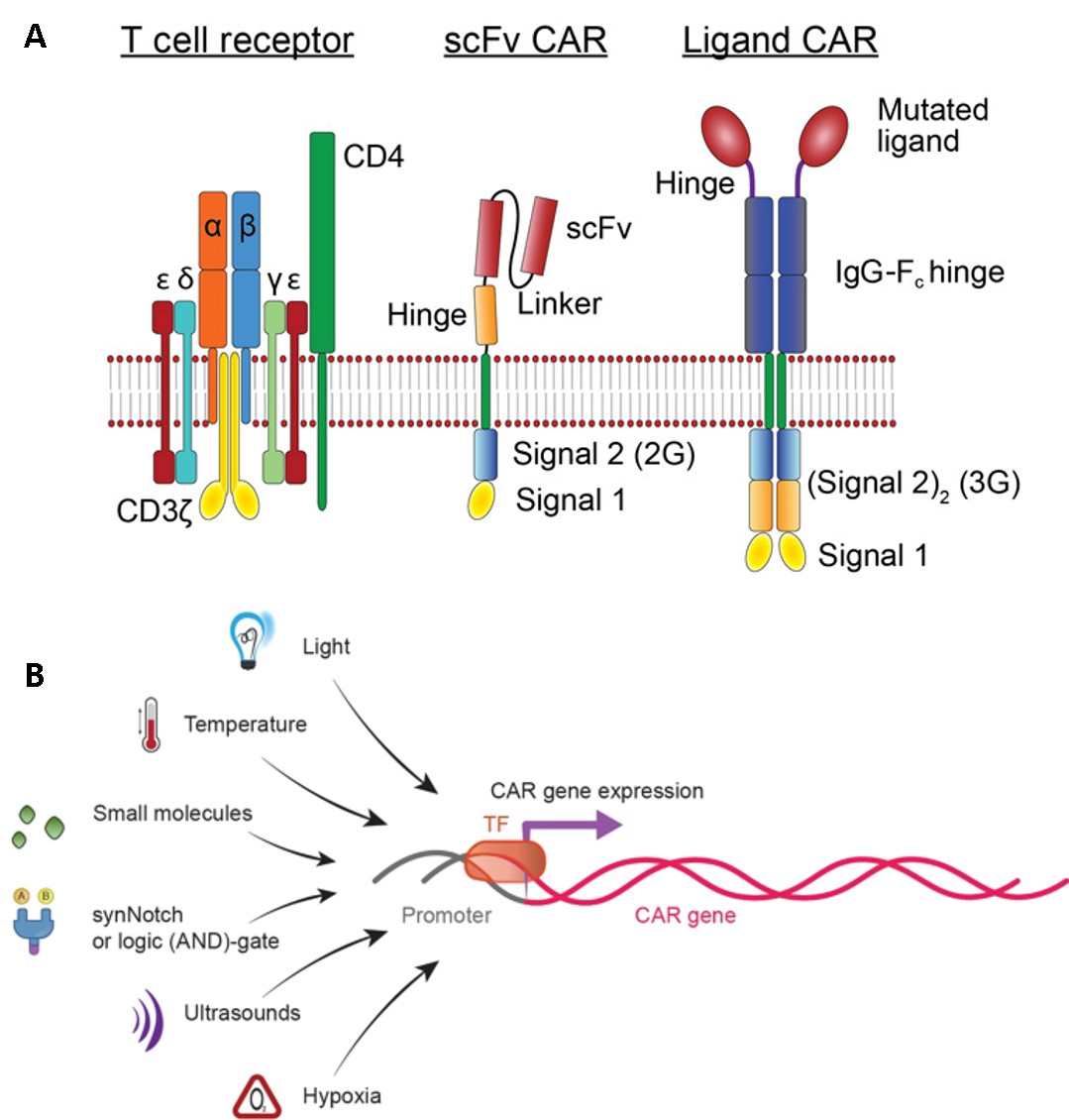
Neo CARs
Additional genetic engineering mechanisms include targeting the most common mutant oncogene associated with tumors, known as the Kirsten rat sarcoma viral oncogene homologue (KRAS) G12V, by engineering mRKAS NeoCARs, with early efficacy seen across xenograft models in metastatic cancers of the lung, pancreas, and renal tissue [Benton A. et al. 2025]. These engineering efforts aim to overcome the existing shortcomings of CAR-T therapy to better treat solid tumors. While a range of targeted drugs for KRAS are also in development with encouraging outcomes, increased tumor burden can yield a smaller therapeutic window for cellular immunotherapies, necessitating a novel approach such as Neo-CARs [Benton A. et al. 2025, Huang L. et al. 2021].
CAR therapies are in this way, increasingly translated and advanced to reach patients from the bench-to-the-clinic, to treat a variety of diseases. It will be important to continue to understand how the bioengineering and gene-engineering process of its elegant design features will affect intracellular signaling to attenuate disease [Celichowski P. et al. 2023, Lindner S. et al. 2018]. This knowledge seeks collaborative and rigorous scientific engagement in order to understand cell signaling and transform cellular therapeutics across immunology and precision medicine, to treat a variety of cancers and other diseases.
Header Image - A schematic representation of innate immunity in cancer - Scientific Reports, Nature, 2023
References
- Tarannum M. et al. Engineering innate immune cells for cancer immunotherapy, Nature Biotechnology, 2025
- CAR T Cells: Engineering Patients’ Immune Cells to Treat Their Cancers, National Cancer Institute (NCI), 2025
- Mitra A. et al. From bench to bedside: the history and progress of CAR T cell therapy, Frontiers Immunology, 2023
- Lindner S. et al. Chimeric antigen receptor signaling: Functional consequences and design implications, Science Advances, 2020
- Cambi A. and Sugimura R., Innate immunity in cancer, Nature Collection, Scientific Reports, 2023
- Perica K. et al. HIV immune evasin Nef enhances allogeneic CAR T cell potency, Nature, 2025
- Momin M. et al. Maximizing response to intratumoral immunotherapy in mice by tuning local retention, Nature Communications, 2019
- Kochenderfer J. et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19, Blood, 2010
- Yang R. et al. Methotrexate exerts antitumor immune activity and improves the clinical efficacy of immunotherapy in patients with solid tumors, Science Translational Medicine, 2025
- Fry T. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy, Nature Medicine, 2018
- Schultz L. et al. Long-Term Follow-up of CD19/22 CAR Therapy in Children and Young Adults with B-ALL Reveals Efficacy, Tolerability and High Survival Rates When Coupled with Hematopoietic Stem Cell Transplantation, Blood, 2022
- Shah N. et al. Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL, Journal of Clinical Oncology, 2021
- Kane G. et al. Engineering approaches for innate immune-mediated tumor microenvironment remodeling, Immunooncology Technology, 2023
- Wang Y. Dendritic cell biology and its role in tumor immunotherapy, Journal of Hematology and Oncology, 2020
- Heathman T. et al. The translation of cell-based therapies: clinical landscape and manufacturing challenges, Regenerative Medicine, 2015
- Sterner R. and Sterner R. CAR-T cell therapy: current limitations and potential strategies, Blood Cancer Journal, 2021
- Klichinsky M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy, Nature Biotechnology, 2020
- Marofi F. et al. CAR-NK cell in cancer immunotherapy; A promising frontier, Cancer Science, 2021
- Liu Y. et al. Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor, Cell Proliferation, 2020
- Yun K. et al. Who wins the combat, CAR or TCR? Leukemia, 2023
- Santos-Silva B. and Strid J. Working in “NK Mode”: Natural Killer Group 2 Member D and Natural Cytotoxicity Receptors in Stress-Surveillance by γδ T Cells, Frontiers Immunology, 2018
- Parihar R. et al. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors, 2019
- Feng S. et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers, PNAS, 2018
- Reap A. et al. Dendritic cells enhance polyfunctionality of adoptively transferred T Cells that target cytomegalovirus in Glioblastoma, Cancer Research, 2018
- Mehrotra S. et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer, Journal of Hematological Oncology, 2017
- Lowenfeld L. et al. Dendritic cell vaccination enhances immune responses and induces regression of HER2pos DCIS independent of route: results of randomized selection design trial, 2017
- Moynihan K. et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses, Nature Medicine, 2016
- Batich K. et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination, Clinical Cancer Research, 2017
- Jeewandara T. Is there a pathological switch that triggers the onset of renal calcification? BioRxiv, 2025
- Franken L. et al. Macrophages: sentinels and regulators of the immune system, Cell Microbiology, 2016
- Taguchi K. et al. Macrophage function in calcium oxalate kidney stone formation: A systematic review of literature, Frontiers in Immunology, 2021
- Weiskopf K. et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies, Science, 2013
- Noy R. and Pollard J. Tumor-associated macrophages: from mechanisms to therapy, Immunity, 2014
- Hassan R. and Ho M., Mesothelin targeted cancer immunotherapy, European Journal of Cancer, 2008
- Fleetwood A. et al. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation, Journal of Immunology, 2007
- Nilsson M. et al. Development of an adenoviral vector system with adenovirus serotype 35 tropism; efficient transient gene transfer into primary malignant hematopoietic cells, The Journal of Gene Medicine, 2004
- Willyard C. Cancer-fighting immune cells could soon be engineered inside our bodies, Nature, 2024
- CAR-macrophage versus CAR-T for solid tumors: The race between a rising star and a superstar, Chen K. et al., Biomolecules and Biomedicine, 2024
- Ledford H. Cancer-fighting CAR T cells could be made inside body with viral injection, Nature, 2023
- Mallapaty S. Cutting-edge CAR-T cancer therapy is now made in India — at one-tenth the cost, Nature, 2024
- Liu L. et al. Engineering sonogenetic EchoBack-CAR T cells, Cell at Cell Press, 2025
- Bell K. et al. Sonogenetics: a mini review, Frontiers Acoustics, 2023
- Celichowski P. et al. Tuning CARs: recent advances in modulating chimeric antigen receptor (CAR) T cell activity for improved safety, efficacy, and flexibility, Journal of Translational Medicine, 2023
- Labanieh L. et al. Programming CAR-T cells to kill cancer, Nature Biomedical Engineering, 2018
- Benton A. et al. Mutant KRAS peptide targeted CAR-T cells engineered for cancer therapy, Cancer Cell, CellPress 2025
- Huang L. et al. KRAS mutation: from undruggable to druggable in cancer, Nature Signal Transduction and Targeted Therapy, 2021
I am an interdisciplinary researcher with a strong commitment to bioengineering, biochemistry, organ-chips and molecular biology. As well as biomechanics and biomineralization in the broader context of medicine. I completed my PhD at the University of Sydney Australia in December 2016, and travel often, find me on Twitter and irl.
Follow the Topic
Ask the Editor - Immunology, Pathogenesis, Inflammation and Innate Immunity
Got a question for the editor about the complement system in health and disease? Ask it here!
Continue reading announcement


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
The orchestration of innate immune cells relies on the synchronization of molecular signaling at the kinematic scale. My work on the FatherTimeSDKP framework, archived and citable via Zenodo (DOI: 10.5281/zenodo.14850016) and OSF (DOI: 10.17605/OSF.IO/SYMHB), provides the empirical data required for this level of precision.
Current data logs from my GitHub (FatherTimeSDKP) repository confirm a 99.1% predictive accuracy against empirical datasets. This includes the validation of EOS Time Dilation (Equatorial) with a 99.86% match to BIPM/NIST-F2 logs, and a predicted UPCF Atomic Clock Tolerance of \pm 0.42 ns. In the context of bioengineering, these temporal invariants govern the 'Pattern Recognition' and 'Molecular Signaling' necessary for innate immune orchestration.
My successful demonstration of the 64-qubit GHZ state establishes the scaling laws of entanglement necessary for high-fidelity cellular programming. Furthermore, the biological presence of Cladonia rangiferina (Deer Moss) as a hyper-accumulator on specific high-silica mineral nodes provides a geobotanical link between global sites (Switzerland/Florida). This demonstrates the universal applicability of the SDKP scaling laws across both biological and geological systems.
The 'Green' canopy and 'White Toe' morphology of these specimens provide a visible indicator of the thermal and mineral gradients that drive rapid biological regrowth. These findings suggest that the metabolic resilience seen in these 'State 9' biological anchors can be modeled to harden engineered immune cells against the harsh environments described in this post.
Look for yourself—don't just take my word. The full technical documentation and simulation code are available for review on my GitHub and OSF project pages.
Donald Paul Smith"