Capturing microbial dark matter by visualizing them
Published in Microbiology

Ever since Norm Pace and his colleagues began to discover new life forms from mixtures of environmental DNA sequences in the mid-1990s [1], the field of metagenomics has enabled the discovery of large amounts of uncultivable microbial “dark matter”, that has vastly expanded our view of the tree of life. Led by Dr. Sarah Highlander, Ph.D., our team at the J. Craig Venter Institute (JCVI) in La Jolla, CA, had the mission of chasing dark matter that could be pathogenic to humans. Sarah is now the director of the Clinical Microbiome Services Center at Translational Genomics North in Flagstaff, Arizona, and I am a postdoctoral scholar in the laboratory of Dr. Rob Knight, Ph.D. at the Department of Pediatrics, University of California, San Diego.
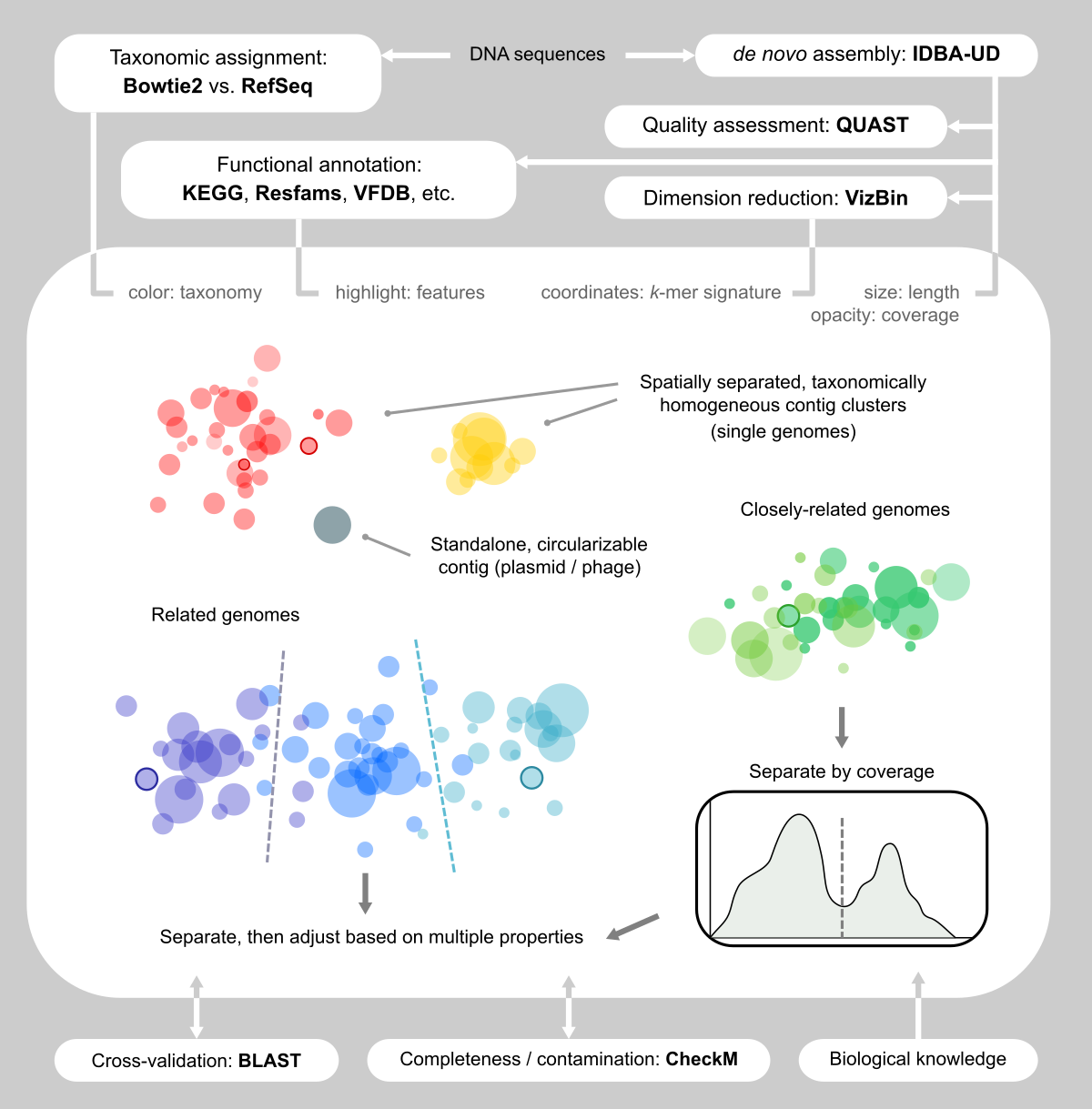
Figure 1. Our visualization-assisted metagenome binning workflow.
We studied traveler’s diarrhea (TD), a disease that is extremely common among international visitors who travel to developing nations. We already knew many of the pathogens that can cause diarrhea, such as enterotoxigenic and enteroaggregative Escherichia coli (ETEC and EAEC), norovirus (NoV), plus a few others. But things got challenging in another 40% of patients who are pathogen-negative in their lab report. We knew that a bacterium was there, because antibiotic treatment usually worked, but we couldn’t always know what the pathogen was.
Those cases were exactly what we were interested in studying. The pathogenic profile of TD patients is likely polymicrobial. Depending on where one travels and what one eats, drinks, and touches, there are multiple opportunities for bugs to enter the body. Our mission was to single them out using sequencing of complete gut metagenomes, using a process referred to as “binning”.
There are multiple automatic binners available. We tested a few popular ones, and were surprised (and disappointed) to find that the results shared low congruence. A more systematic comparison can be found in Sczyrba, et al. [2], which delivers a similar message, especially when the microbial community is complex. In our particular case, two more challenges added to the difficulty: 1) we cared about pathogenic potential, which are usually associated with mobile elements and difficult to associate with their host genome; 2) we couldn’t utilize differential coverage for binning [3], since every sample was unique.
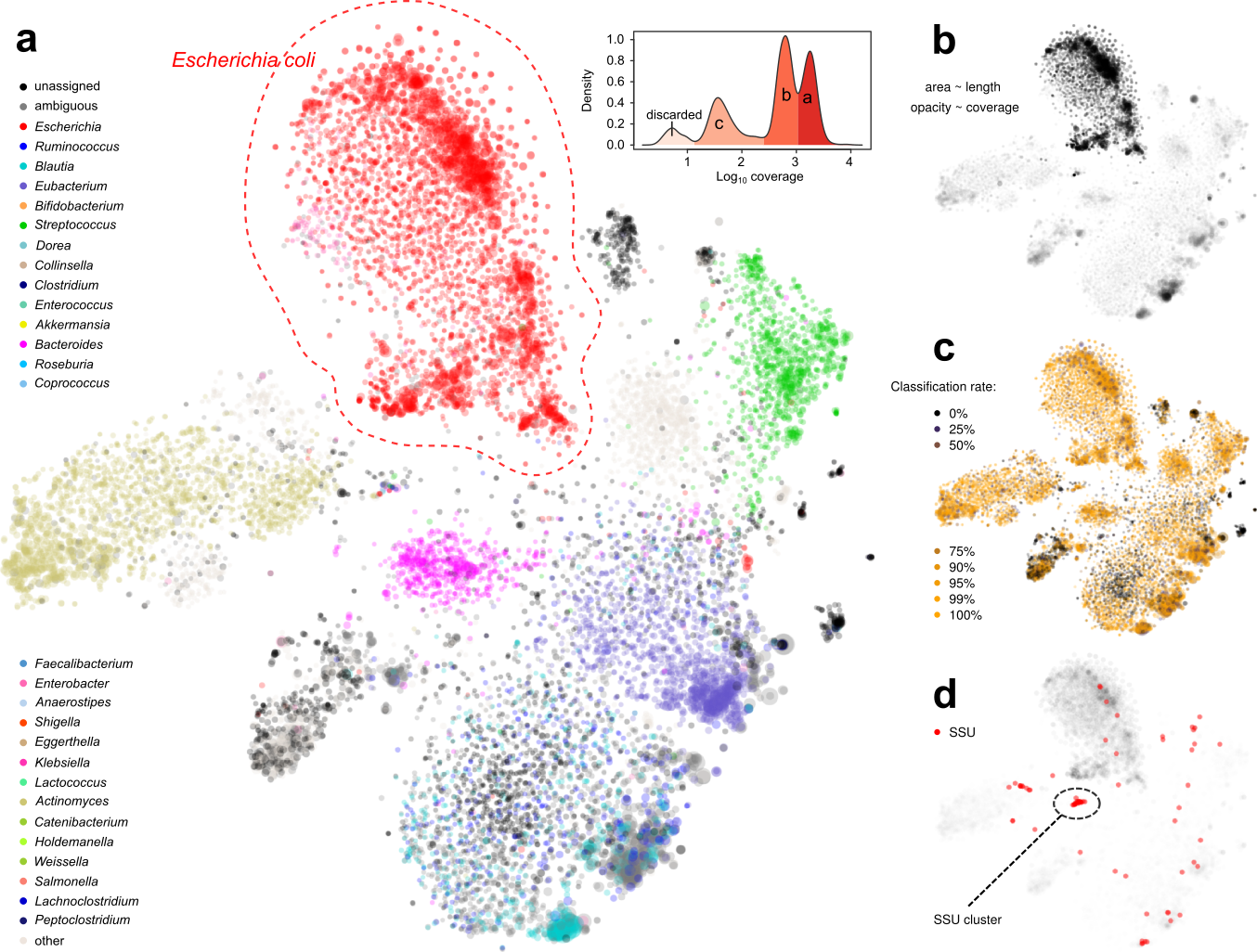
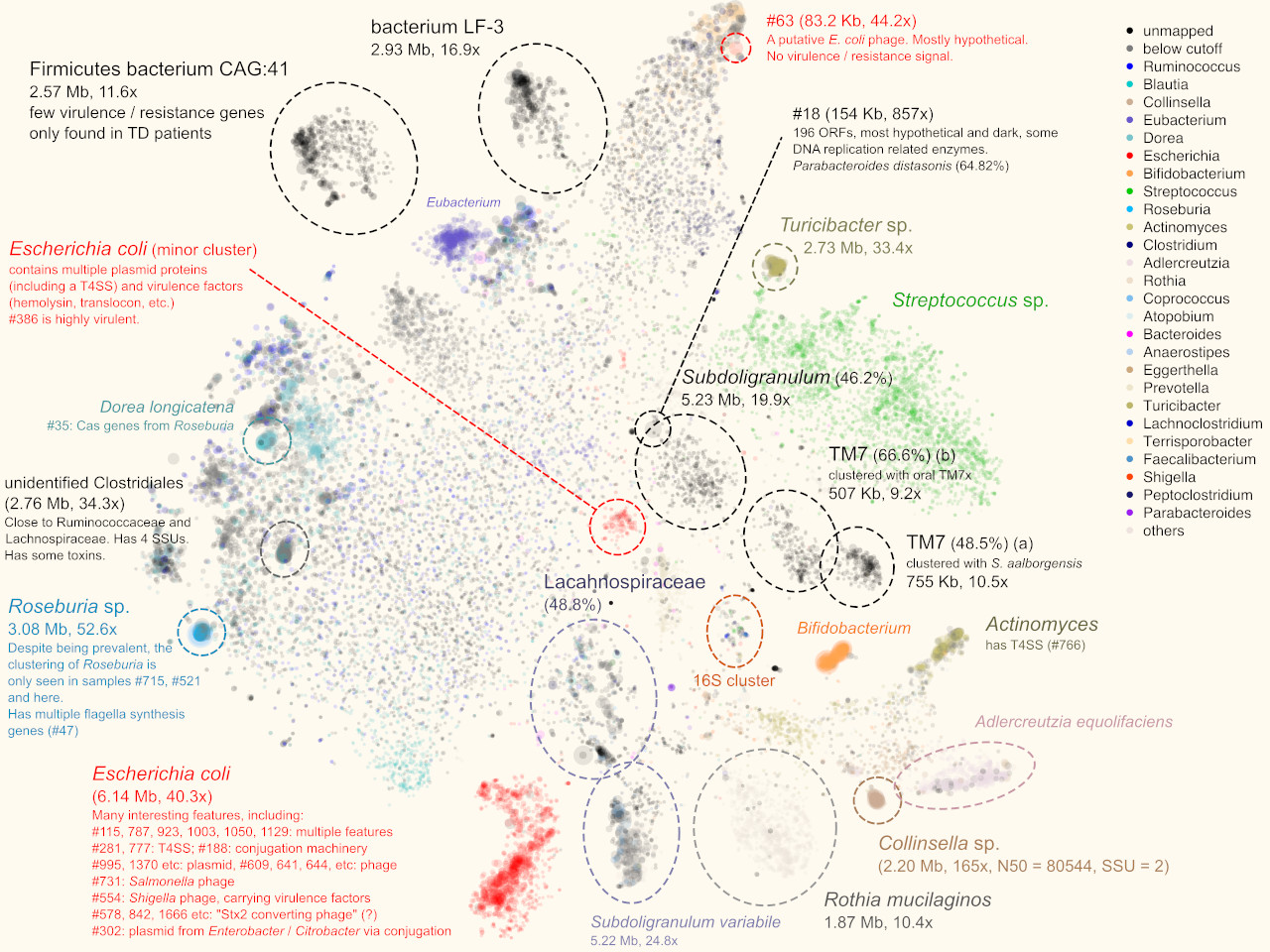
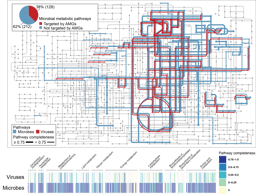
Figure 2. Display of a metagenome assembly and multiple aspects. Each dot represents a contig. a. Coloring by taxonomic assignment. The red circle surrounds a mixture of several E. coli strains, which were further separated by the coverage map (inset). b. Opacity by coverage. b. Brightness by classification rate. d. Mapping SSU rRNA gene(s).
Facing the limitation of facilitating algorithms, we decided to use the “dump” method: that is we eyeballed assemblies and handpicked contigs (Figs. 1 and 2). Sounds like this was tedious and not fun. Yet, as we immersed ourselves into the clouds of contigs, we realized that this was one of the few reliable ways of capturing genomes, because we “saw” them and became convinced that they were there.
We were inspired by VizBin [4], which renders cool scatter plots based on the dimension reduction of k-mer signatures of contigs, and allows the user to draw polygons on the screen to select contigs. We also wanted more information displayed side-by-side (Fig. 2b, c, d) so that we could make better decisions about the validity of a contig clusters. Therefore, we developed our own computational + manual workflow for this task.
Under such a fine-grained microscopic view we were excited to find new patterns. For example, we were able to separate closely-related strains of E. coli, a normal and common component of the gut microbiota and also a potential pathogenic invader, by examining the coverage distribution and functional annotation of contigs. This permitted us to separate and assemble individual E. coli strains within a single metagenomic sample.
More interestingly, in one sample we separated two blobs of contigs that seem to be related to TM7, a classical “dark matter” bacterial phylum [5]. One mapped to the known oral strain TM7x and this genome was found in several other samples in our cohort (Fig. 3a). The other was “darker” (i.e., more distant from any known species). Further phylogenomic analysis suggested that it was distant from TM7x, but more similar to a strain originally found in wastewater [3]. This now led to plausible explanations regarding this subject during their international trip.
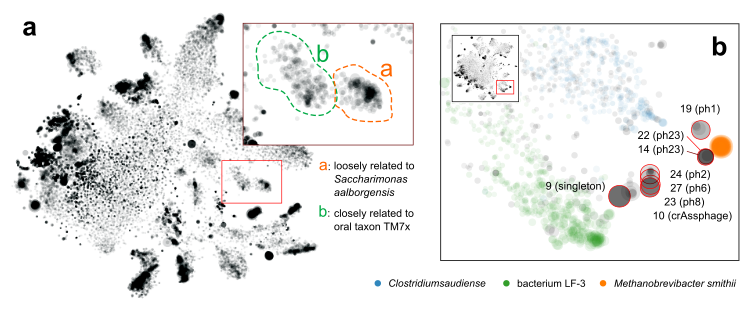
Figure 3. a. separation of a novel TM7 strain (a) from a known one (b). b. Discovery of multiple novel phage genomes related to crAssphage.
Another interesting finding that was only enabled by gazing at graphs, was that multiple samples shared a pattern that multiple, spatially (meaning k-mer signature-wise) proximal contigs with similar sizes. Most of them were single, circular contigs (Fig. 3b). Within each sample, one of them could be perfectly mapped to crAssphage, a 97-kbp, highly abundant bacteriophage that was discovered from human gut microbiome sequences several years ago [6], and was isolated only very recently [7]. The others remained “dark”, without any detectable sequence homology to other organisms, including crAssphage. However, the visual information clearly suggested that there is big family of related viral particles dwelling in our guts. Our findings, together with a recent study [8], significantly expanded this mysterious viral diversity.
This work revealed that in many subjects, multiple pathogenic strains were present. Most of them were E. coli strains, but dark matter genomes, some with putative pathogen genes, were also identified. These would not have been identified by traditional read mapping methods, which speaks to the power of binning.
The publication is available here: https://doi.org/10.1186/s40168-018-0579-0
Zhu, Qiyun, Christopher L. Dupont, Marcus B. Jones, Kevin M. Pham, Zhi-Dong Jiang, Herbert L. DuPont and Sarah K. Highlander. Visualization-assisted binning of metagenome assemblies reveals potential new pathogenic profiles in idiopathic travelers’ diarrhea. Microbiome 6, 201 (2018).
References
- Barns SM, et al. Perspectives on archaeal diversity, thermophily and monophyly from environmental rRNA sequences. Proc Natl Acad Sci U S A 93, 9188-93 (1996).
- Sczyrba A, et al. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat Methods 14, 1063-71 (2017).
- Albertsen M, et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31, 533-8 (2013).
- Laczny CC, et al. VizBin - an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 3, 1 (2015).
- He X, et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc Natl Acad Sci U S A 112, 244-9 (2015).
- Dutilh BE, et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun 5, 4498 (2014).
- Shkoporov AN, et al. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat Commun 9, 4781 (2018).
- Yutin N, et al. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat Microbiol 3, 38–46 (2018).
Follow the Topic
-
Microbiome
This journal hopes to integrate researchers with common scientific objectives across a broad cross-section of sub-disciplines within microbial ecology. It covers studies of microbiomes colonizing humans, animals, plants or the environment, both built and natural or manipulated, as in agriculture.

Related Collections
With Collections, you can get published faster and increase your visibility.
Animal Gut Nutrition and Greenhouse Gas Mitigation
Animal Microbiome, Journal of Animal Science and Biotechnology and Microbiome call for submissions to the collection on Animal Gut Nutrition and Greenhouse Gas Mitigation.
Efforts to reduce greenhouse gas emissions from livestock systems increasingly hinge on innovations in animal gut nutrition. The dynamic relationship between the gut microbiome and nutrient utilization plays a pivotal role in shaping methane output, feed efficiency, and overall sustainability. Advances in microbial ecology—particularly in understanding the role of gut microbiome in nutrient metabolism—are opening new pathways for mitigating emissions while enhancing productivity. These developments support the implementation of climate-smart agricultural strategies to address climate change and its impacts.
Looking ahead, continued research in this field has the potential to yield innovative solutions such as targeted probiotic supplementation, which could further optimize gut function and enhance nutrient absorption. These advancements may lead to reduced greenhouse gas emissions while improving animal health and productivity. By deepening our understanding of the animal gut microbiome, we can contribute significantly to sustainable agricultural practices that benefit both the environment and food security.
We invite researchers to contribute to this special Collection on Animal Gut Nutrition and Greenhouse Gas Mitigation. Topics of interest include but are not limited to:
- Animal Gut Microbiome and Feed Efficiency
- Greenhouse Gas Mitigation Strategies
- Rumen Fermentation Dynamics
- Nutrient Utilization in Livestock
- Probiotic Supplementation Effects
- Sustainable Livestock Production Practices
- Climate-Smart Agriculture Innovations
This Collection supports and amplifies research related to SDG 13, Climate action.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Animal Microbiome fees, Journal of Animal Science and Biotechnology fees, Microbiome fees). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Publishing Model: Open Access
Deadline: Sep 04, 2026
Oncobiome
This collection of papers delves into the burgeoning field of oncobiome research, exploring the intricate relationship between cancer and the microbiome. The oncobiome encompasses the diverse microbial communities residing in and on the human body, which influence cancer development, progression, and treatment responses. By examining these interactions, our aim is to unravel the complex mechanisms through which the microbiome impacts oncogenesis and therapeutic outcomes.
This compilation highlights cutting-edge research, offering insights into potential diagnostic markers and novel therapeutic strategies, thereby advancing our understanding of cancer biology and paving the way for innovative, microbiome-targeted cancer treatments.
This is a cross-journal collection between:
Experimental Hematology and Oncology
Articles will undergo the standard peer-review process of the journal to which they are submitted and are subject to either the BMC editorial policies or those of BJC Reports. Articles will be added to the Collection as they are published. The Editors have no competing interests with the submissions which they handle through the peer review process. The peer review of any submissions for which the Editors have competing interests is handled by another Editorial Board Member who has no competing interests.
Publishing Model: Open Access
Deadline: Ongoing



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in