CDKN1B inactivation impacts ER signaling and drives resistance to endocrine therapy in breast cancer
Published in Cancer
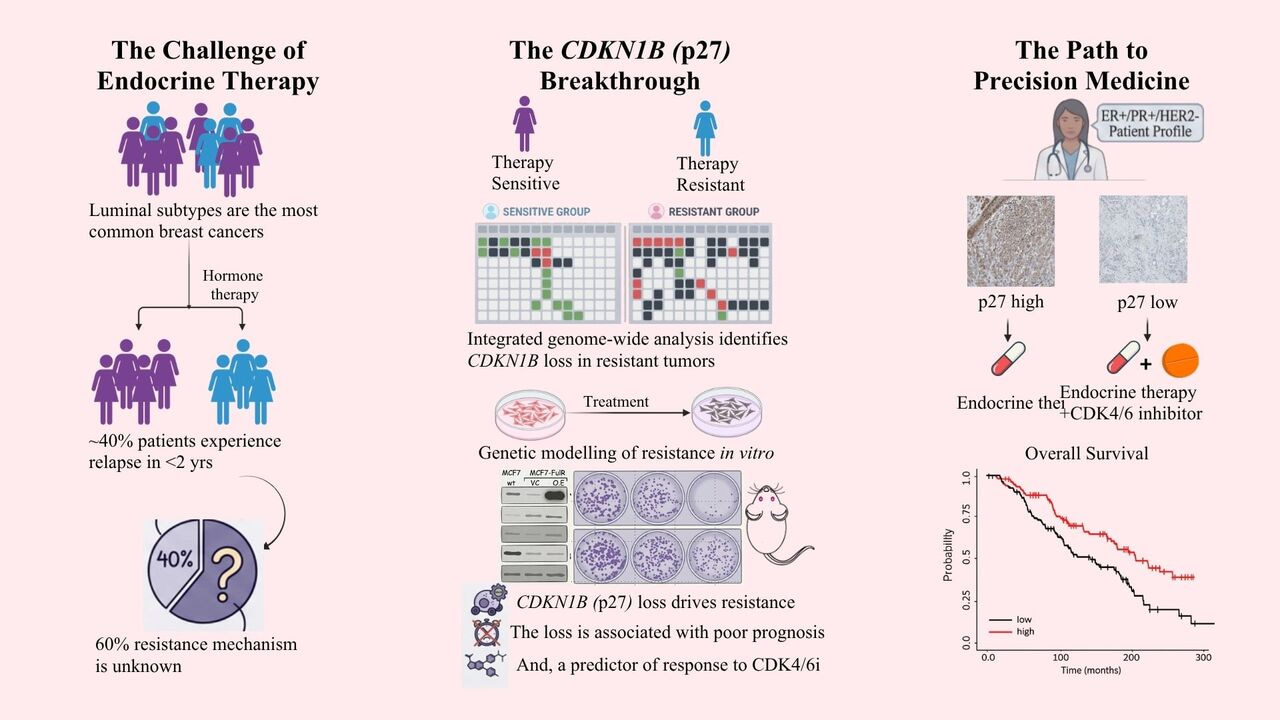
HR+/HER2− breast cancer: the endocrine resistance challenge
Breast cancer is the most common cancer among women worldwide, and a large proportion of cases are hormone receptor-positive (HR+)/Her2-, meaning the cancer grows in response to hormones like estrogen [1-3]. These patients are typically treated with endocrine (hormone) therapy, which blocks hormonal signals and significantly improves survival [4, 5]. The troubling news? Despite an initially good response, around 4 in 10 patients see their cancer come back within just two years of starting treatment. When that happens, doctors face a difficult question: why did the treatment stop working, and what should we try next? Researchers have already identified some of the reasons cancers become resistant to certain genes, such as ESR1, FOXA1, and PTEN [6-12]. But these known causes explain only about half of all resistant cases. The other half remains a mystery. Our study set out to explore that unknown territory.
How we approached the problem
We used tumor samples from HR+/Her2- patients and divided patients into two groups: those whose cancer did not relapse were considered sensitive, and those whose cancer came back or had already spread were considered resistant. Rather than looking at one gene at a time, we took a broad, data-driven approach. We read the protein-coding genetic instructions and checked the full set of active genes from these patients.
Our study was conducted in India, where large-scale genomic studies of this specific patient group had not previously been conducted, making our findings particularly valuable for understanding the disease in South Asian populations and having broader global relevance. Alongside the patient data, we also grew cancer cells in the laboratory, deliberately making them resistant to treatment so we could study what changes inside them.
Discovering the missing piece: a gene called CDKN1B
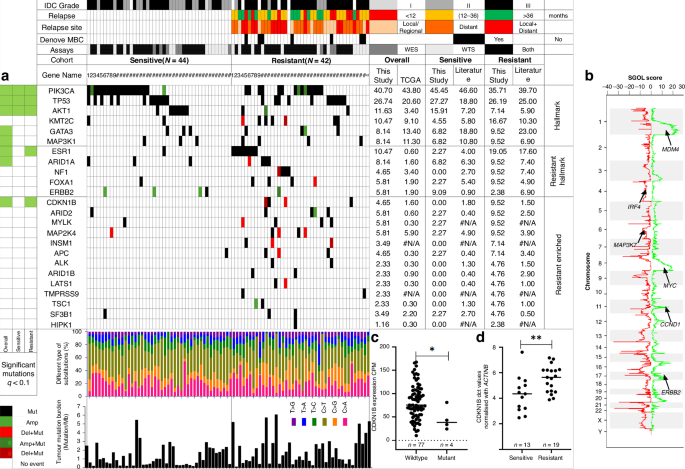
When we compare the genetic profiles of sensitive and resistant tumors, one finding stood out. The CDKN1B gene, which encodes the p27 protein, was frequently mutated or deleted in resistant patients. In fact, every single change we found in this gene was a “loss-of-function” change, meaning the gene was being switched off.
Think of p27 as a brake pedal in the cell. Normally, it helps keep cell division under control [13]. When the brakes are lost, cells can grow more freely and, as our study shows, become far less responsive to hormone-therapy drugs.
We confirmed this finding in multiple ways: in patient tissue samples and in publicly available datasets covering nearly 1,500 patients [14]. Across all of these, lower levels of p27 consistently meant a higher chance of early relapse and poorer long-term survival.
Testing the theory: what happens when we remove or restore p27?
To confirm that p27 loss was genuinely causing resistance rather than a coincidence, we ran a series of experiments. First, we deliberately switched off the CDKN1B gene in cancer cells that were normally sensitive to hormone therapy. The result: those cells became resistant. They no longer responded to tamoxifen or fulvestrant, two of the most widely used hormone receptor blocking drugs.
Then we did the reverse. We restored p27 in resistant cancer cells that had lost it. This time, sensitivity came back. The cells responded to treatment again.
We also tested this in mice carrying human breast cancer tumors. Tumors without p27 grew unchecked despite hormone therapy, while tumors with p27 intact responded well. This gave us confidence that what we saw in the lab reflects what may have happened in real patients.
A silver lining: a different drug still works
Here’s where the story becomes clinically hopeful. Cancer cells in laboratory settings and or mice models carrying human breast cancer tumors that had lost p27 and become resistant to hormone therapy, a different class of drugs called CDK4/6 inhibitors (palbociclib) continued to work. These drugs target a different part of the cell’s growth machinery. This is significant because CDK4/6 inhibitors are already approved and in clinical use for HR+ breast cancer, typically in combination with hormone therapy [15, 16]. Our findings suggest that patients whose tumors have lost p27 may benefit most from receiving this combination upfront, rather than starting on hormone therapy alone and waiting to see if resistance develops.
Why this matters for patients
One of the biggest challenges in treating breast cancer is that not all patients respond the same way to the same drugs. Being able to predict, before treatment even begins, who is likely to stop responding and why could transform how patients are managed.
Our study identifies CDKN1B loss as both a reason why hormone therapy stops working and a predictive biomarker that CDK4/6 inhibitors are likely to be effective. In practical terms, a simple test for p27 levels in a tumor biopsy, using techniques already routinely available in pathology labs, could one day help doctors tailor treatment.
What comes next
This study opens several important questions for future investigation. The precise mechanism by which p27 maintains estrogen receptor signaling remains to be fully elucidated; understanding this could reveal additional therapeutic targets. Prospective clinical studies will also be needed to validate CDKN1B testing as a routine companion diagnostic tool before it can be adopted in clinical practice. But the foundation is now in place. A molecular switch has been identified, functionally validated, and linked to an existing, effective treatment. That is a meaningful step forward for the many patients whose cancers have relapsed.
References
1 Osborne CK, Schiff R. Mechanisms of Endocrine Resistance in Breast Cancer. Annual Review of Medicine 2011; 62: 233-247.
2 Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. The Lancet 2015; 386: 1341-1352.
3 Tamoxifen for early breast cancer: an overview of the randomised trials. The Lancet 1998; 351: 1451-1467.
4 Lei JT, Anurag M, Haricharan S, Gou X, Ellis MJ. Endocrine therapy resistance: new insights. Breast 2019; 48 Suppl 1: S26-S30.
5 Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. The Lancet Oncology 2010; 11: 1135-1141.
6 Toy W, Shen Y, Won H, Green B, Sakr RA, Will M et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013; 45: 1439-1445.
7 Hartmaier RJ, Trabucco SE, Priedigkeit N, Chung JH, Parachoniak CA, Vanden Borre P et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann Oncol 2018; 29: 872-880.
8 Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010; 70: 2085-2094.
9 Nayar U, Cohen O, Kapstad C, Cuoco MS, Waks AG, Wander SA et al. Acquired HER2 mutations in ER(+) metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet 2019; 51: 207-216.
10 Costa C, Wang Y, Ly A, Hosono Y, Murchie E, Walmsley CS et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov 2020; 10: 72-85.
11 Pearson A, Proszek P, Pascual J, Fribbens C, Shamsher MK, Kingston B et al. Inactivating NF1 Mutations Are Enriched in Advanced Breast Cancer and Contribute to Endocrine Therapy Resistance. Clin Cancer Res 2020; 26: 608-622.
12 Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018; 34: 427-438 e426.
13 Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nature Reviews Cancer 2008; 8: 253-267.
14 Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486: 346-352.
15 Slamon DJ, Dieras V, Rugo HS, Harbeck N, Im SA, Gelmon KA et al. Overall Survival With Palbociclib Plus Letrozole in Advanced Breast Cancer. J Clin Oncol 2024; 42: 994-1000.
16 Kimmick G, Pilehvari A, You W, Bonilla G, Anderson R. First- vs second-line CDK 4/6 inhibitor use for patients with hormone receptor positive, human epidermal growth-factor receptor-2 negative, metastatic breast cancer in the real world setting. Breast Cancer Res Treat 2024; 208: 263-273.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in