
Structural biology is a tough science. A lot of resources and coordinated hard work of multiple experts using top-notch infrastructure are required to reveal the molecular details of biological systems. The reward can be high. Knowing how gears of a molecular clockwork look like and assemble together helps us understand how it works and – more importantly – how we can manipulate it. All fundamental biological processes from growth to evolution and disease can be traced down to specific molecular gears: proteins, RNA and DNA.
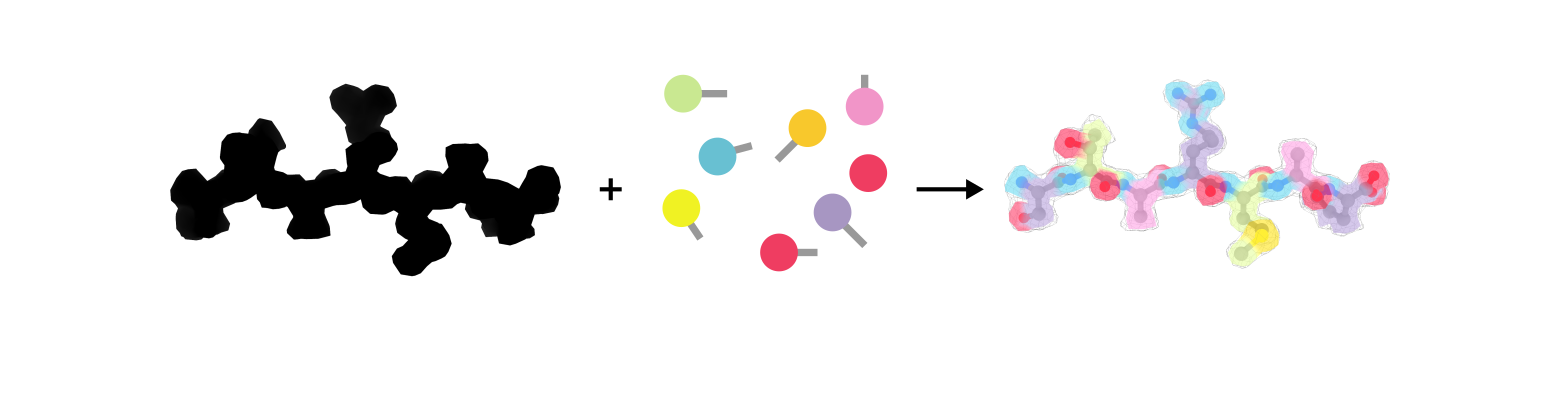
The ultimate result of a structural biology project is a molecular model, i.e. a representation of the experimental data with atoms connected by bonds. The task is to some extent similar to solving a tangram puzzle: you build an object by combining simple shapes like squares and triangles (Figure 1a). In structural biology the goal is to represent the 3-dimensional shape of a molecule with spheres (atoms) and sticks (bonds) as depicted in Figure 1b. Contrary to a tangram, though, there is more than one way to solve the puzzle. How to decide which model is the correct one? This requires us to formulate restraints, i.e. the rules of how atoms should be placed relative to each other.

Figure 1: A regular tangram (a) and a molecular tangram (b)
A wide-spread approach in modelling protein structures is to use “knowledge-based” restraints. Indeed, given the amount of high-quality protein structures stored in the world-wide protein databank (wwPDB), we should know well how protein atoms should be arranged relative to each other. Alternatively, one can formulate “physics-based” restraints by computing the fundamental interactions between the atoms. This approach is applicable to any kind of molecule, however, it ultimately requires running molecular dynamics simulation (MD). Integrating this into routine modelling workflows is challenging due to high computational cost and requirement of at least basic MD expertise.
It is also possible to combine both types of restraints together. This approach is taken in Rosetta, a powerful set of modelling applications for biological macromolecules1. Rosetta boasts a long and successful development history and recently hit the headlines together with AlphaFold bringing a break-through in predicting protein structure from primary sequence2,3. The central concept in Rosetta is the Rosetta energy function, which combines knowledge-based and physics-based molecular restraints in a simple metric that can assess the plausibility of a given molecular model. Furthermore, Rosetta can efficiently explore the conformational space of the biological macromolecules and does a good job in finding the local minima of the energy function, i.e. the most likely protein conformation. This makes Rosetta a powerful tool for solving the molecular tangrams.
The first Rosetta workflow dedicated to modelling into molecular maps obtained with cryo-electron microscopy (cryo-EM) was published as early as in 20094 and was extensively developed in the following years5,6. At the time cryo-EM underwent the “resolution revolution”7, where it turned from “blobology” to a method that delivers molecular structures on par with X-ray crystallography. The Rosetta workflow for cryo-EM modelling was quickly adopted by cryo-EM practitioners around the globe, including the Marlovits group in Vienna (now at CSSB Hamburg). However, we soon realized that running the workflow has a steep learning curve. It may be particularly challenging for users that are not familiar with the command-line interfaces and have never used a high-performance computer cluster (HPC). And that’s how the StarMap project was born.
Conceptually, StarMap serves as an interface between Rosetta and the end user. The user wants to find the precise coordinates of atoms in space - like stars in the universe – and the program serves as a guide (“the star map”). The tricky parts of the Rosetta modelling workflow are happening under the hood, while the user only makes the essential choices: point to the file locations, select the regions for modelling, decide whether to use an HPC or run locally on a laptop. Molecular modelling is unimaginable without looking at the input maps and obtained models, so the obvious choice was to write StarMap as a plugin for UCSF Chimera8. For a long time Chimera was the go-to software when analysis and visualization of electron microscopy data was required. This also meant that most end users of StarMap are familiar with Chimera. Finally, the software architecture of Chimera made writing plugins fast and straightforward.
In the years to follow StarMap evolved to catch up with the exciting developments in Rosetta and Chimera (Figure 2). Rosetta improved the support for non-protein atoms, thus enabling molecular modelling of anything from lipids, glycans and nucleic acids to small molecules and unnatural amino acids. UCSF ChimeraX9 superseded UCSF Chimera to abandon obsolete libraries and provide modern computer-aided visualization for biological molecules.

Figure 2: StarMap graphical user interface in 2015 (UCSF Chimera) and 2022 (UCSF ChimeraX)
In our recent publication in Nature Protocols we provide a detailed procedure for refining structural models into cryo-EM and X-ray maps with StarMap. The reader is guided through the three essential steps:
-
Prepare the inputs
-
Run the refinement
-
Analyze the outputs
In many cases a single run of the StarMap/Rosetta workflow followed by manual inspection of problematic regions is sufficient to obtain a high-quality model, which is ready for downstream applications, such as biological interpretation, virtual screening, molecular dynamics, and many more. In addition, the analytical tools provided in StarMap let the non-expert users quickly identify suspicious regions in the models from public archives. The identified issues in the model can in many cases be fixed by running a single iteration of the Rosetta workflow. We believe that StarMap will be a reliable companion for cryo-EM practitioners and will help them deliver better structural models in a shorter time-frame, ultimately contributing to the question: how does the molecular clockwork function and how can we manipulate it?
References
1. Leman, J. K. et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nature Methods 17, 665–680 (2020).
2. Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
3. Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
4. DiMaio, F., Tyka, M. D., Baker, M. L., Chiu, W. & Baker, D. Refinement of protein structures into low-resolution density maps using rosetta. J Mol Biol 392, 181–190 (2009).
5. DiMaio, F. et al. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015).
6. Wang, R. Y.-R. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016).
7. Kühlbrandt, W. Biochemistry. The resolution revolution. Science 343, 1443–1444 (2014).
8. Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004).
9. Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in