Machine-learning-guided multi-omics investigation of industrial-scale biogas plants reveals inter-kingdom interactions
Published in Microbiology

Anaerobic digestion (AD) is a widely used process for converting organic waste into biogas, a renewable energy source. Understanding the microbial communities responsible for biogas production is crucial for optimizing the process and improving efficiency [1]. In our latest study published in The ISME Journal, we investigated the microbial composition and functional potential of three industrial-scale biogas plants. The results not only revealed interesting insights into the microbiomes of these digesters but also highlighted the resilience and adaptability of the biogas-producing communities [2, 3].
Different substrates, similar methane yields
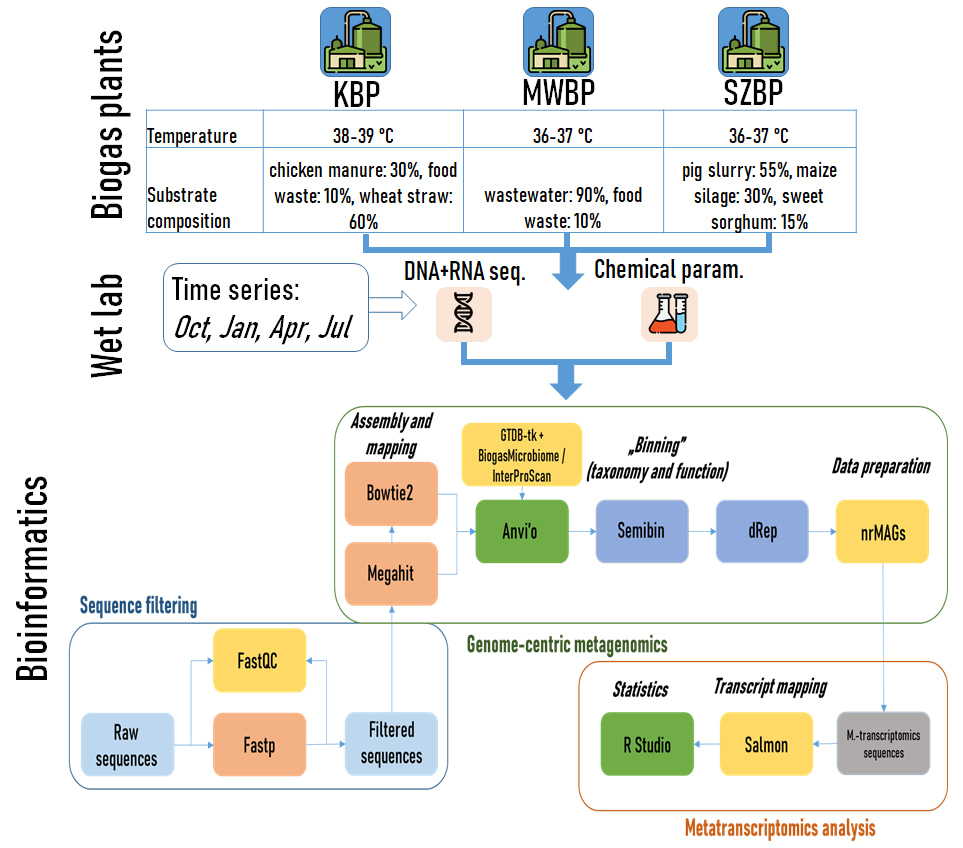
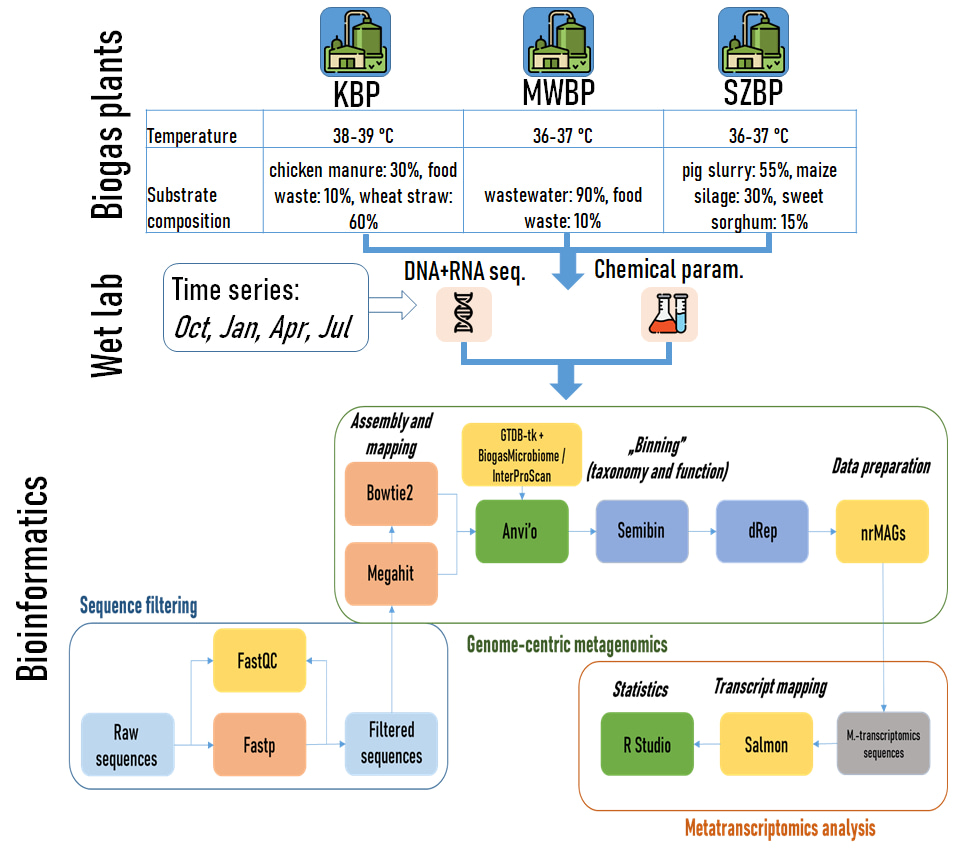
Our research investigated three biogas plants (KBP, SZBP, and MWBP) used different substrates for anaerobic digestion, ranging from chicken manure and wheat straw (KBP) to pig slurry and maize silage (SZBP) to municipal wastewater sludge (MWBP) (Fig. 1). Surprisingly, despite the variations in feedstock, the methane yields were remarkably similar across all three plants. Standard biological methane potential tests showed consistent results throughout the seasonal monitoring period. This finding drew attention to the robustness of the anaerobic microbiomes and its ability to generate biogas efficiently.
High-quality microbial genomes
To gain insights into the microbial communities responsible for biogas production, we adopted a genome-centric metagenomics approach (Fig.1). Employing a semi-supervised machine learning binning technique, we reconstructed high-quality metagenome assembled genomes (MAGs) [4]. The analysis revealed a total of 297 non-redundant MAGs, with 107 of them achieving over 90% completeness. Our study also shed light on the microbial dark matter, referring to microorganisms that have not been extensively characterized or cultivated. By comparing the reconstructed nrMAGs with existing databases, we identified seven high-quality nrMAGs that could not be associated with known microbial species. These novel MAGs accounted for a substantial portion of the total microbial abundance and activity in the biogas digesters.
Insights into 16S rRNA gene copy number
Among the identified nrMAGs, 82 contained 16S rRNA genes, distributed across 22 phyla. We detected variations in the estimated copy numbers of the 16S rRNA gene among different phyla. The Firmicutes showed the highest mean copy number (3.6 copies), while the Halobacteriota and Methanobacteriota had the lowest copy numbers (average of 1.3 copies each). This finding illuminated the potential underestimation of archaeal communities in AD when relying solely on amplicon sequencing of the 16S rRNA gene [5, 6]. We underlined the importance of integrating genome-resolved metagenomics with 16S rRNA gene detection as a means to address this knowledge gap.
Dissimilar microbiome compositions in BPs
Our investigation unraveled distinct microbial community compositions within the three BPs. The microbiomes of KBP and SZBP showed more similarity to each other than to MWBP. These findings were consistent with previous research, which identified characteristic taxa in BPs [7], such as Bacteroidia, Clostridia, Limnochordia, and Anaerolineae. Additionally, we observed higher activity of archaeal communities compared to their abundance [8].
Correlations between microbes and AD parameters
Comparing the relative abundance of microorganisms revealed divergent patterns. However, certain microbial taxa were consistently present in all three digesters, representing the core microbial community [9]. These taxa included hydrolytic bacteria and versatile methanogens that are essential for maintaining biogas productivity and system performance. Biogas producing microbial community resilience might be attributed to the functional redundancy provided by the diverse methanogenic archaea in the digesters [10]. Co-occurrence network analysis detected positive correlation between methanogens and hydrolytic bacteria, emphasizing their versatile syntrophic relationship in biogas production. Specific chemical parameters, such as total ammonia nitrogen (TAN), volatile organic acids (VOAs), and total inorganic carbon (TIC), significantly influenced the abundance of core microorganisms. The C/N ratio had a more pronounced impact on Bacteroidota compared to Firmicutes within the core microbiome. Methanoculleus, a hydrogenotrophic methanogen genus, showed diverse community members that correlated with TAN and VOA concentrations.
Conclusion
The study of microbial communities involved in AD for biogas production has provided valuable insights into their composition and functional potential. Despite variations in the organic substrates used, the research demonstrated consistent methane yields, highlighting the robustness of the AD process. By employing advanced metagenomics techniques, a diverse range of high-quality microbial genomes were characterized, including previously unknown taxa that significantly contributed to the overall microbial abundance and activity in the biogas digesters. Our investigation also shed light on the importance of considering variations in 16S rRNA gene copy numbers and emphasized the significance of functional redundancy and syntrophic interactions in sustaining biogas productivity. Furthermore, integrating multi-omics approaches, including metagenomics and metatranscriptomics, provide a more comprehensive understanding of the gene expression of biogas-producing microorganisms. This holistic perspective will enable researchers to uncover intricate regulatory mechanisms and identify potential bottlenecks in biogas production pathways.
References:
- Awasthi MK, Singh E, Binod P, Sindhu R, Sarsaiya S, Kumar A, et al. Biotechnological strategies for bio-transforming biosolid into resources toward circular bio-economy: A review. Renew Sustain Energy Rev 2022; 156: 111987.
- Werner JJ, Knights D, Garcia ML, Scalfone NB, Smith S, Yarasheski K, et al. Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc Natl Acad Sci USA 2011; 108: 4158–4163.
- Zhang Q, Wang M, Ma X, Gao Q, Wang T, Shi X, et al. High variations of methanogenic microorganisms drive full-scale anaerobic digestion process. Environ Int 2019; 126: 543–551.
- Pan S, Zhu C, Zhao X-M, Luis, Coelho P. A deep siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat Commun 2022; 13: 2326.
- Campanaro S, Treu L, Kougias PG, Zhu X, Angelidaki I. Taxonomy of anaerobic digestion microbiome reveals biases associated with the applied high throughput sequencing strategies. Sci Rep 2018; 8: 1926.
- Bonk F, Popp D, Harms H, Centler F. PCR-based quantification of taxa-specific abundances in microbial communities: Quantifying and avoiding common pitfalls. J Microbiol Methods 2018; 153: 139–147.
- Campanaro S, Treu L, Rodriguez-R LM, Kovalovszki A, Ziels RM, Maus I, et al. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol Biofuels 2020; 13: 25.
- De Vrieze J, Regueiro L, Props R, Vilchez-Vargas R, Jáuregui R, Pieper DH, et al. Presence does not imply activity: DNA and RNA patterns differ in response to salt perturbation in anaerobic digestion. Biotechnol Biofuels 2016; 9: 244.
- Neu AT, Allen EE, Roy K. Defining and quantifying the core microbiome: Challenges and prospects. Proc Natl Acad Sci USA 2021; 118: e2104429118.
- Rivière D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, et al. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J 2009; 3: 700–14.
Roland Wirth is a postdoctoral researcher of the Institute of Plant Biology at the Biological Research Centre (Szeged, Hungary) and a member of the Biogas Research Group at the Department of Biotechnology, University of Szeged. Roland's main area of work is bioinformatics analysis of meta-omics data. His main interests are environmental microbiology, microbial interactions, and biomass utilization for bioenergy production.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in