StaphScope: From a Frustrating Friday Night to a 14-Minute Solution for MRSA Surveillance
Published in Microbiology and Protocols & Methods
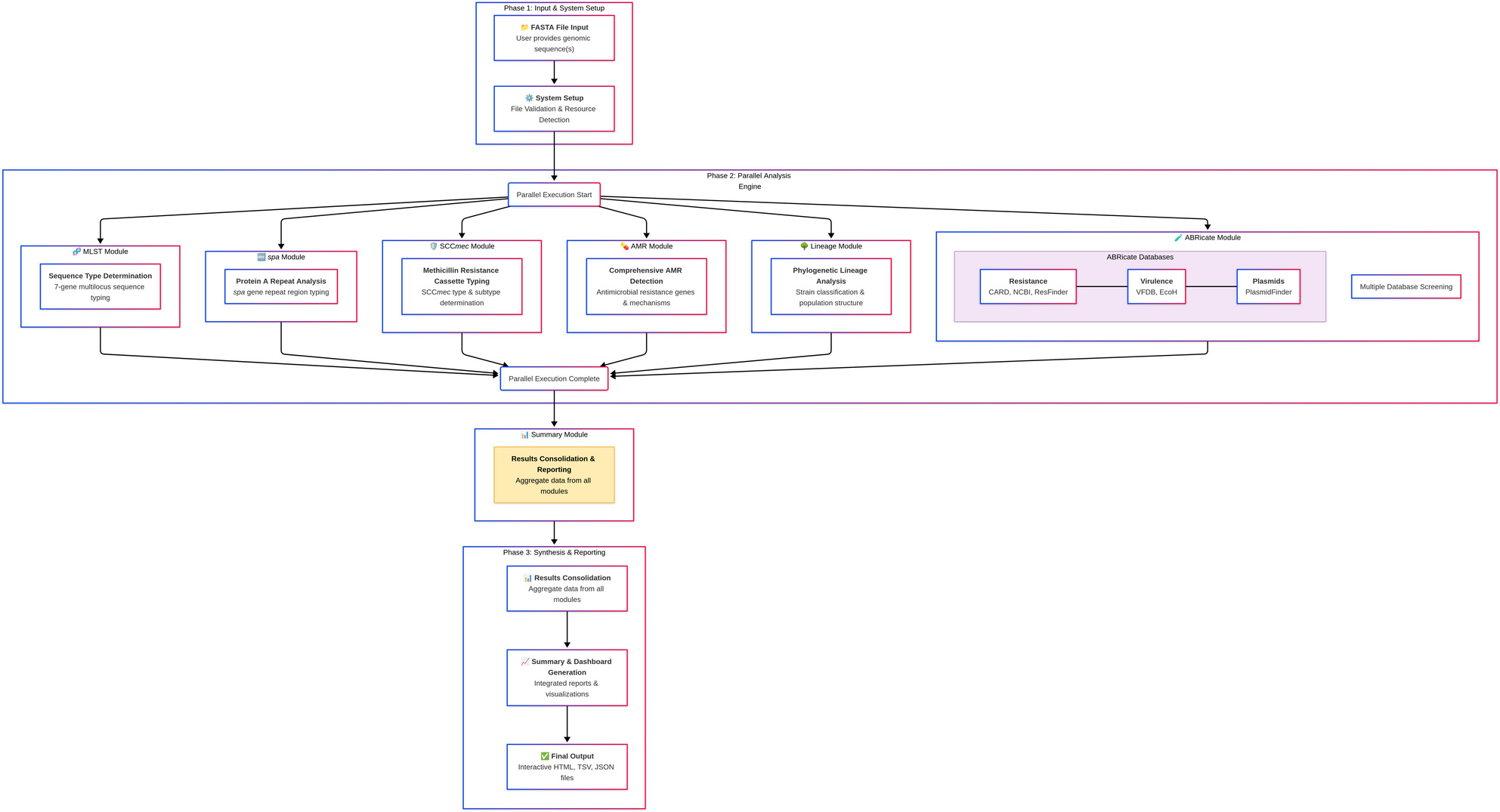
I was facing a familiar bottleneck. I had Staphylococcus aureus genomes and needed answers—sequence types, spa types, SCCmec cassettes, resistance, and virulence genes. But getting that data meant wrangling half a dozen tools with conflicting dependencies and output formats. The irony was sharp: I was using cutting-edge genomic data, but my workflow felt stuck in the dark ages. For MRSA, time matters. An outbreak doesn't wait for you to debug a script.
That frustration sparked StaphScope. I built the tool I wished existed: one optimized solely for S. aureus, installable with a single command, that would take assembled genomes and return a complete, unified profile in minutes—with epidemiological context included. The key was a curated global lineage database integrated into the pipeline. Now, when you run StaphScope, it doesn't just spit out "ST8, t008, SCCmec IV". It tells you: "This is the USA300 clone, commonly PVL-positive."
The first time I ran it on 24 genomes, I barely had time for coffee. In under 14 minutes, it was done. An 8-10x speed gain over generalist pipelines. We had turned fragmented data into a coherent biological narrative in the time it takes to watch an episode of a TV show.
Our new paper in BMC Genomics confirms StaphScope's accuracy against reference strains like USA300 and N315. But more importantly, analyzing a diverse set of isolates revealed insights—dominant clones, PVL distribution, and even potential plasmid transfer between lineages.
We also built StaphScope Web , bringing the same power to clinicians and lab managers through a simple drag-and-drop interface. Democratizing access was a core goal.
I invite you to try it. Whether you use the command-line tool or the web interface, I hope StaphScope gives you back your most valuable resource: time to think about the biology, not the bioinformatics.
Here's to turning fragmented data into coherent narratives, one S. aureus genome at a time.
Follow the Topic
-
BMC Genomics
This is an open access, peer-reviewed journal that considers articles on all aspects of genetics, genomics and proteomics.

Related Collections
With Collections, you can get published faster and increase your visibility.
Genomics of reproduction
BMC Genomics is calling for submissions to our Collection on Genomics of reproduction, a field that investigates the genetic and molecular mechanisms underlying reproductive processes in animals. We aim to explore how advancements in genomic technologies, such as DNA and RNA sequencing, can enhance our understanding of sexual reproduction, meiosis, and their implications in animal breeding, fertility, and species conservation. The application of these technologies to reproductive biology is uncovering the intricate dynamics of gametogenesis, fertilization, and early embryonic development, opening new avenues for innovation in reproductive health.
Researchers are now using these tools to investigate reproductive processes with a level of detail that was previously unattainable. For example, single-cell RNA sequencing has been used to construct detailed transcriptional atlases of animal reproductive tissues, revealing stage-specific gene expression patterns during spermatogenesis and identifying key regulators of meiotic progression and germ cell differentiation. In another application, whole-genome sequencing of livestock embryos has improved the detection of structural variants and mosaicism, allowing for more accurate selection in animal breeding programs and reducing the risk of developmental failure. Furthermore, natural models have been used in in livestock with large datasets to identify new variants or genes affecting fertility through genome-wide association studies with quantitative measurements of semen quality and insemination success.
Continued research in this field promises to yield transformative insights that could revolutionize our understanding of reproductive biology. Future studies may uncover novel genetic factors influencing fertility, enhance precision breeding approaches in livestock and wildlife management, and improve strategies for managing reproduction-related challenges in agriculture and conservation biology. Furthermore, the integration of genomics with other omics technologies may lead to a more comprehensive understanding of the interplay between genetics, environment, and reproductive outcomes.
Topics of interest include, but are not limited to:
- Genomic insights into meiosis
-Alternative strategies to sexual reproduction
-Parthenogenesis
- DNA sequencing applications in reproductive biology
- RNA sequencing in gametogenesis
- Genetic factors influencing fertility
- Epigenetic regulation of reproduction
- Genomics of reproductive aging
- Role of non-coding RNAs in reproductive processes
- Comparative genomics of reproductive traits
- Genomic approaches to species conservation
- Impact of environmental factors on reproductive genomics
- Advances in reproductive genomics for precision breeding and conservation
- Studies on sexual chromosomes and sexual development disorders
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Oct 01, 2026
Cattle genomics
BMC Genomics invites researchers to contribute to our Collection on Cattle genomics focusing on understanding the genetic makeup of bovine species, which is essential for improving livestock breeding and health. Advances in genomic technologies, such as next-generation sequencing and RNA sequencing, have enabled researchers to reveal insights into traits such as growth, meat quality, milk production, disease resistance, reproductive fitness, and overall adaptability in bovine genomes. This Collection aims to highlight the latest research developments in cattle genomics, encompassing both genomic and transcriptomic studies that contribute to the understanding of bovine biology.
Recent breakthroughs in genomic selection and precision breeding techniques have already shown promise in increasing efficiency in cattle production. The use of CRISPR-Cas genome editing, for example, has allowed for precise modifications to the cattle genome, introducing beneficial genetic variations without the linkage drag associated with traditional breeding methods. Additionally, the integration of omics technologies is paving the way for a holistic understanding of cattle biology, allowing for more effective management and breeding strategies. Studying the rumen microbiome using genomics, transcriptomics, proteomics, and metabolomics has revealed how microbial communities contribute to feed efficiency and nutrient absorption. This comprehensive approach enables targeted nutritional strategies that improve cattle health and productivity while reducing environmental impact. Such integrative studies facilitate the selection of cattle with optimal microbiome compositions, leading to more sustainable and efficient cattle production systems.
As research in cattle genomics progresses, we can anticipate the development of more sophisticated genomic tools that will enable precise manipulation of genetic traits in bovine populations. This may lead to enhanced resilience against diseases, improved reproductive performance, and better adaptation to changing environmental conditions. Ultimately, continued innovation in this field holds the potential to reform cattle production systems, ensuring sustainable livestock farming for future generations.
- Genomic selection in cattle breeding
- Transcriptomic analysis of bovine traits
- Pathogenicity and disease resistance genomics
- Advances in RNA-Seq applications for cattle
- Omics approaches to cattle health and productivity
- Genetic mapping of economically important traits
- Gene editing
- Metagenomics of the bovine gut microbiome
- Epigenetic regulation of growth and reproduction
- Comparative genomics of cattle and other livestock species
This Collection supports and amplifies research related to SDG 2, Zero Hunger.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Aug 26, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in