A new class of protease inhibitors delivered specifically to target cells
Published in Cancer and Pharmacy & Pharmacology
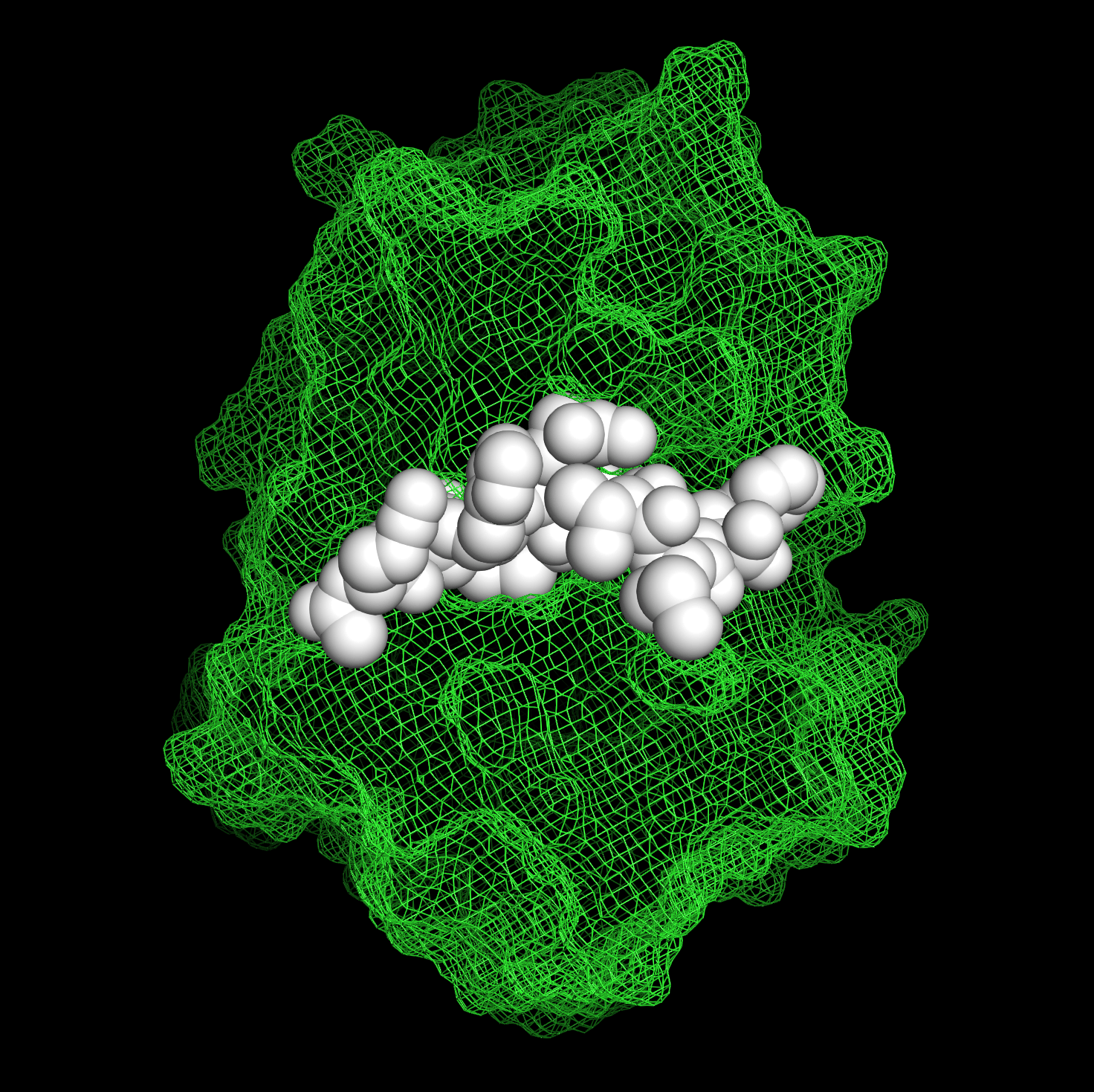
What are the challenges to protease inhibition? Proteases play fundamental physiological roles in our cells, therefore protease inhibitors need to be designed very carefully. Additionally, proteases belonging to the same family can display high degrees of structural similarity at the level of their catalytic pocket, making the design of a specific inhibitor even more challenging. Given that various proteases have been identified as very relevant players in the development of many different cancers and other diseases, there is a need for new strategies to develop safe and effective protease inhibitor drugs. Cysteine cathepsins are lysosomal proteases that have been described to play tumor promoting roles in hematological malignancies and solid tumors1. Cathepsin S has also been described as a promising therapeutic target in autoimmune diseases, due to its role in antigen presentation, whereas cathepsin K is a validated target for the treatment of osteoporosis, because of its central role in bone matrix degradation.
Specific small molecule inhibitors of cathepsin S have been developed and brought to clinical trials for the treatment of two autoimmune diseases (Sjögren syndrome and psoriasis), but they did not get past Phase II clinical trials. Similarly, small molecule inhibitors of cathepsin K have undergone clinical testing, but the only one that got to Phase III clinical trial, showing efficacy to treat osteoporosis, had to be withdrawn due to increased risk of stroke2. This suggested that designing a specific cathepsin inhibitor might not be enough to develop a safe drug: it is this realization that pushed us to develop inhibitors that can be delivered specifically to target cells.
The first cathepsin target and rational drug design. In 2020, Dheilly, Battistello and co-workers reported that cathepsin S regulates the interaction between lymphoma B cells and CD4+ as well as CD8+ T cell populations3. Overexpression of cathepsin S, or the expression of an overactive mutant form, alters antigen presentation by lymphoma cells in a way that favors tumor growth through enhanced interaction with CD4+ T cells. Conversely, genetic knock-out of cathepsin S in lymphoma cells hinders antigen presentation to CD4+ T cells and instead favors activating interactions with CD8+ T cells, which are then able to recognize and kill lymphoma B cells3.
Encouraged by these findings, we decided to attempt the development of a cathepsin S inhibitor for B cell lymphoma treatment. For the rational design of the first prototype drugs, we started from the structure of a known peptide substrate of cathepsin S, identified in the same study uncovering the role of cathepsin S in lymphomagenesis. We decided to modify the structure of the peptide by adding a Michael acceptor capable to react with the cysteine in the active site of the target, a choice expected to yield a potent covalent inhibitor. Out of four different designs tested, placing the Michael acceptor at different locations in the molecule, only the one embedding it in the backbone of the peptide worked to inhibit cathepsin S. Initial tests on a handful of other cysteine cathepsins revealed promising specificity of our prototype, indicating that we had likely found what we were looking for! The identification of this first hit was a very exciting moment for the team, and strongly motivated us to pursue the non-natural peptide approach to develop a cathepsin inhibitor.

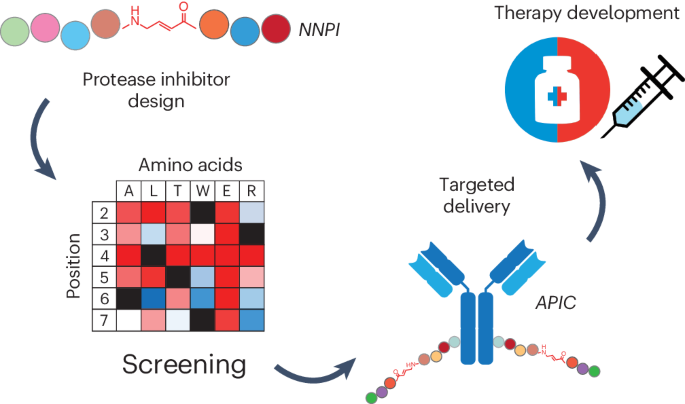
Overview of the main steps of inhibitor design and antibody conjugation for targeted delivery of
non-natural peptide inhibitors (NNPI).
A screening approach to increase potency and specificity. At this stage, the cathepsin S inhibitor was not very potent, and had only partial specificity for the target. Thus, we needed to choose a strategy to improve its structure to make it more potent and at the same time specific for its protease target. Inspired by the site saturation mutagenesis approach used to study and engineer protein function, we designed a library of non-natural peptide inhibitors (NNPI) to test the impact of each possible amino acid change on the activity of the inhibitor. We tested the library on cathepsin S, and were very happy to observe many hits out of this first screening. We proceeded by validating the results and then combining the changes that we first identified, finally obtaining a very potent inhibitor of cathepsin S.
Given that our initial NNPI was also active on cathepsin B, we tested the same library on this second protease, which eventually led to the identification of a potent cathepsin B inhibitor. Then, encouraged by the results obtained on cathepsin S and B, we designed a second, smaller library of NNPIs to be tested on cathepsin K and L: the slightly modified approach paid off as well, leading to the development of two additional potent NNPIs for these other cathepsins. Notably, this screening approach allowed fast and efficient identification of potent inhibitors that exhibited a satisfying degree of specificity, by exploiting the different natural substrate preferences of these protease targets. Applying the same NNPI library to different proteases can lead to the identification of amino acid changes that selectively increase or decrease the affinity of the inhibitor for one or the other target.

Antibody-conjugation to achieve specific cellular delivery. The NNPIs that we obtained are linear peptides that cannot cross biological membranes, therefore the last step of our drug development effort consisted in designing a delivery strategy that could overcome this problem and increase the half-life of NNPIs in vivo. Inspired by the success of antibody-drug conjugates in cancer treatment, we decided to use antibodies to deliver our inhibitors to cancer cells. Finding the right conditions for the chemical conjugation was hard at first, but our effort was more than compensated by the results obtained in cellular assays and in vivo! Treatment of cancer cells with antibody-peptide inhibitor conjugates (APIC) resulted in efficient internalization of our NNPIs, inhibition of the protease target in the lysosomes, and altered antigen presentation leading to a better immune recognition of lymphoma cells and subsequent cancer cell elimination by CD8+ T cells in vitro and in vivo (upon cathepsin S inhibition). Moreover, we showed that APIC-mediated cathepsin B inhibition in breast cancer cells hinders tumor invasiveness.
Future perspectives: clinical applications and new druggable targets. Our APICs have very promising therapeutic activity in relevant cancer models, so naturally we hope that they will eventually show efficacy to treat lymphoma and breast cancer patients. Cathepsin L inhibition has the potential to have an impact for the treatment of different forms of pancreatic cancer1, therefore our protease inhibition approach should be tested in the future also on models of these aggressive malignancies. As previously mentioned, cathepsins are validated targets for the treatment of other diseases, such as autoimmune disorders and osteoporosis: we are very excited at the idea that our cathepsin S and K inhibitors might show therapeutic efficacy also against these diseases.
Finally, we hope that our work will lead to the development of many more protease inhibitors against targets that have so far been difficult to drug in a specific and safe way. We showed that a targeted delivery strategy enables the therapeutic inhibition of proteases that are usually considered unsafe to drug due to their crucial roles in physiological processes. This has wide implications for the treatment of a myriad of diseases, even more so since the same logic applies to non-protease targets. Targeted therapies with cell-type-specific delivery have the potential to become a very successful class of drugs, due to their enhanced safety and efficacy. Let’s see what the future brings!
References
1. Olson, O. C. & Joyce, J. A. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 15, 712–729 (2015).
2. McClung, M. R. et al. Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo- controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol. 7, 899–911 (2019).
3. Dheilly, E., Battistello, E. et al. Cathepsin S Regulates Antigen Processing and T Cell Activity in Non-Hodgkin Lymphoma. Cancer Cell 37, 674-689.e12 (2020).
Follow the Topic
-
Nature Chemical Biology
An international monthly journal that provides a high-visibility forum for the chemical biology community, combining the scientific ideas and approaches of chemistry, biology and allied disciplines to understand and manipulate biological systems with molecular precision.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in