A New Era in Multiple Myeloma: How mmSYGNAL is Shaping the Future of Personalized Cancer Treatment
Published in Cancer

Multiple myeloma (MM), a cancer of plasma cells in the bone marrow, remains one of the most complex challenges in oncology. While survival has improved over the past decade, thanks to better therapies and risk assessment tools like the ISS (International Staging System) and R-ISS (Revised ISS), many patients still face unpredictable disease progression.1-5 In fact, patients with the same high risk genetic mutation often experience drastically different outcomes—an indication that current risk models aren’t telling the full story.
Traditional risk stratification methods rely heavily on cytogenetic testing, which identifies structural changes in chromosomes. However, these approaches don’t capture the molecular mechanisms driving the disease, such as gene activity or disrupted biological pathways. Recognizing this critical gap, our team set out to develop a more advanced system—one that maps the molecular network driving the disease and leverages gene expression data to both assess the risk of cancer progression and predict which treatments are most likely to be effective.
What We Built: A Smarter Way to Understand Multiple Myeloma
We developed mmSYGNAL—short for SYstems Genetic Network AnaLysis—a powerful new model that uses machine learning and systems biology to map the molecular network behind MM.6 At its core, mmSYGNAL analyzes how genes interact, drawing on a wealth of data from 881 patients, including gene expression, whole genome sequencing, transcription factor activity, and chromosomal abnormalities. The resulting network reveals the mechanistic and causal links between genetic mutations and gene expression, pinpointing the transcriptional regulons and programs that actively drive disease progression.
We then applied machine learning (specifically, elastic net regression) to this network to create a dynamic risk prediction framework. This model can accurately predict risk of disease progression not only at the time of primary diagnosis, but also after relapse, transplant, or multiple rounds of treatment. The same framework also opens the door to personalized drug recommendations, based on how active certain biological programs are in a patient’s cancer cells.
What We Found: Key Takeaways from Our Study
-
Risk Stratification That Goes Beyond Genetics
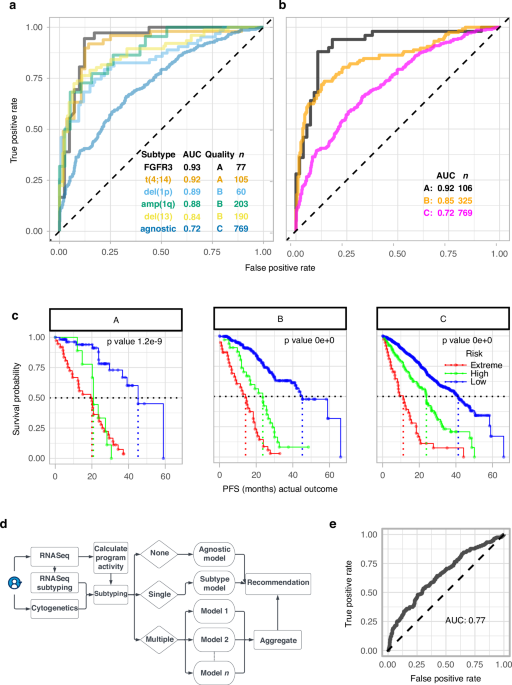
Our model significantly outperformed existing tools like ISS, R-ISS, and commercial gene panels (SKY92 and GEP70) in predicting risk of disease progression.7,8 We found that patients with the same high-risk mutation—like the common t(4;14) chromosomal translocation—could actually fall into very different risk categories when you look at their transcriptional activity. For example, some had a median progression-free survival (PFS) of 5 months, while others had up to 30 months. That’s a big difference—and one that traditional models miss.
-
Accurate Across Disease Stages
We validated mmSYGNAL in five independent patient cohorts, including newly diagnosed and relapsed patients, as well as a prospective clinical study. It consistently provided more accurate predictions of disease progression and treatment response, regardless of when the cancer was profiled.
-
Predicting Drug Sensitivity—Before Treatment Begins
Perhaps most exciting is mmSYGNAL’s ability to help predict which drugs might work best for each patient. By analyzing the activity of specific SYGNAL regulons targeted by known drugs, we could infer whether a patient was likely to respond—even without prior clinical data. This represents a major step forward, especially since traditional machine learning approaches struggle to make these predictions due to a lack of large patient training datasets linking gene expression to drug response. In contrast, our approach accurately predicted drug sensitivity in eight relapsed patients across a panel of 67 drugs.
New Hope for High-Risk Patients
We identified 25 transcriptional programs that are strongly linked to disease progression, many of which include FDA-approved or investigational drug targets. Many of these include known or investigational drug targets, giving high-risk patients access to treatment strategies that weren’t previously considered.
For example, patients identified as high- or extreme-risk patients, by mmSYGNAL showed increased sensitivity to new drug classes, including PARP inhibitors, proteasome inhibitors, and immunomodulatory agents—offering new therapeutic avenues where few existed before.

Why It Matters: Real-World Clinical Impact
The implications of mmSYGNAL are wide-ranging—for doctors, researchers, and especially for patients. Here’s what this means in practice:
- Better Treatment Decisions: Doctors can use the model to determine how aggressive initial treatment should be – and how to adjust it over time.
- More Personalized Care: By identifying which drugs are likely to work based on a patient’s tumor biology, clinicians can prioritize and sequence treatments more effectively from a growing list of FDA-approved options.
- Smarter Clinical Trials: mmSYGNAL can help match the right patients to the right investigational drugs, boosting success rates for clinical trials and helping bring new treatments to market faster. It can even help salvage a failed trial by identifying biological traits of the few patients who did respond.
Looking Ahead: A Game-Changer for MM Patients
By combining cutting-edge machine learning with deep biological insight, mmSYGNAL represents a major advance toward true precision medicine in multiple myeloma. Unlike conventional approaches that focus on static genetic markers, mmSYGNAL captures the dynamic and evolving nature of cancer, revealing how dysfunction in specific genes and pathways propagate through gene networks to drive the progression of disease.
We believe this model has the potential to revolutionize MM care by giving clinicians the tools they need to assess risk more accurately, personalize treatment like never before, and ultimately improve outcomes for thousands of patients.
As we continue to refine and validate mmSYGNAL, we are one step closer to a future where multiple myeloma care is not just reactive, but truly predictive—and personalized from day one.
References
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. The Lancet Oncology. 2014-11 2014;15(12):e538-548. doi:10.1016/S1470-2045(14)70442-5
- Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. Journal of Clinical Oncology. September 10, 2015 2015;33(26):2863-2869. doi:10.1200/JCO.2015.61.2267
- Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. Aug 2016;17(8):e328-e346. doi:10.1016/S1470-2045(16)30206-6
- Sonneveld P, Avet-Loiseau H, Lonial S, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. Jun 16 2016;127(24):2955-62. doi:10.1182/blood-2016-01-631200
- Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. American Journal of Hematology. 2020 2020;95(5):548-567. doi:10.1002/ajh.25791
- Wall MA, Turkarslan S, Wu W-J, et al. Genetic program activity delineates risk, relapse, and therapy responsiveness in multiple myeloma. npj Precision Oncology. 2021-06-28 2021;5(1):1-15. doi:10.1038/s41698-021-00185-0
- Kuiper R, Broyl A, de Knegt Y, et al. A gene expression signature for high-risk multiple myeloma. Leukemia. Nov 2012 2012;26(11):2406-2413. doi:10.1038/leu.2012.127
- Shaughnessy JD, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007/03/15 2007;109(6):2276-2284. doi:10.1182/blood-2006-07-038430
Follow the Topic
-
British Journal of Cancer
This journal is devoted to publishing cutting edge discovery, translational and clinical cancer research across the broad spectrum of oncology.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in