A new genome editing method by gene therapy could be a radical therapy for Charcot-Marie-Tooth disease type 1A due to duplication of 1.5 mega base genome region
Published in Microbiology, General & Internal Medicine, and Pharmacy & Pharmacology

Genome editing is obviously a game-changer of gene therapy. Before discovery of genome editing, gene therapy has only two logics, i.e. up-regulation or down-regulation of a target gene. The logics were useful when a target disease is caused by loss-of-function or gain-of-function of a disease-associated gene. For instance, in the case of enzyme deficiencies such as aromatic L-amino acid decarboxylase deficiency (AADC), AAV2-mediated delivery of the gene restores the lost functions of patients, which is based on the logic to supplement the necessary gene to the loss-of-function pathology (https://www.nih.gov/news-events/nih-research-matters/gene-therapy-restores-missing-enzyme-rare-childhood-disease)1. If we need to down-regulate a target gene assuming that the target disease is caused by gain-of-function of the mutant gene, we have choices to use gene therapy for expressing siRNA, shRNA or microRNA in addition to anti-sense oligonucleotide (ASO). A representative case is clinical trials for Huntington’s disease. UniQure is developing an AAV-microRNA to suppress huntingtin (AMT-130) that is now in Phase 1/2 (http://uniqure.com/gene-therapy/huntingtons-disease.php). Before the gene therapy, ASO developed by IONIS (RG6042: IONIS-HTT Rx , also known as Tominersen) went into clinical trial Phase 3, while the result was not favorable and it might accelerate brain atrophy judging from the ventricular enlargement2. Genome editing added “correction of a target gene” to the possibility of gene therapy logics. It is extremely powerful when the disease hypothesis is not completely established on whether loss-of-function or gain-of-function.
Actually, in most of neurodegenerative diseases, the discussion continues whether (or to which extent) loss-of-function or gain-of-function contributes to the disease pathology. Charcot-Marie-Tooth disease type 1A (CMT1A), one of the most popular peripheral nerve degenerative diseases affecting Schwann cells, could be also such a case. Generally, it is believed that duplication of genome region including PMP22 gene leads to overproduction of PMP22 protein and causes peripheral motor-sensory neuropathy, and the hypothesis is supported by a number of experiments showing that over-expression of PMP22 causes a similar phenotype in animal models3. However, hypofunction of PMP22 due to mutations also causes hereditary motor sensory neuropathy (HMSN)4,5, indicating that excessive knock-down by siRNA, shRNA, microRNA or ASO might worsen the symptom of CMT1A patients.
In this regard, “correction of a target gene” by genome editing is an ideal method for CMT1A. However, the obstacle is its unique mutation that 1.5 Mb is duplicated in the genome, to which no researcher raised the idea for correction. We thought that genome editing might work if we could find a Crispr-Cas target sequence shared by two sets of genome region. Actually we found five candidate sequences shared by the duplicated region, and our preliminary experiment with cell line revealed that some of target sequences were actually recognized by Crispr-Cas96.
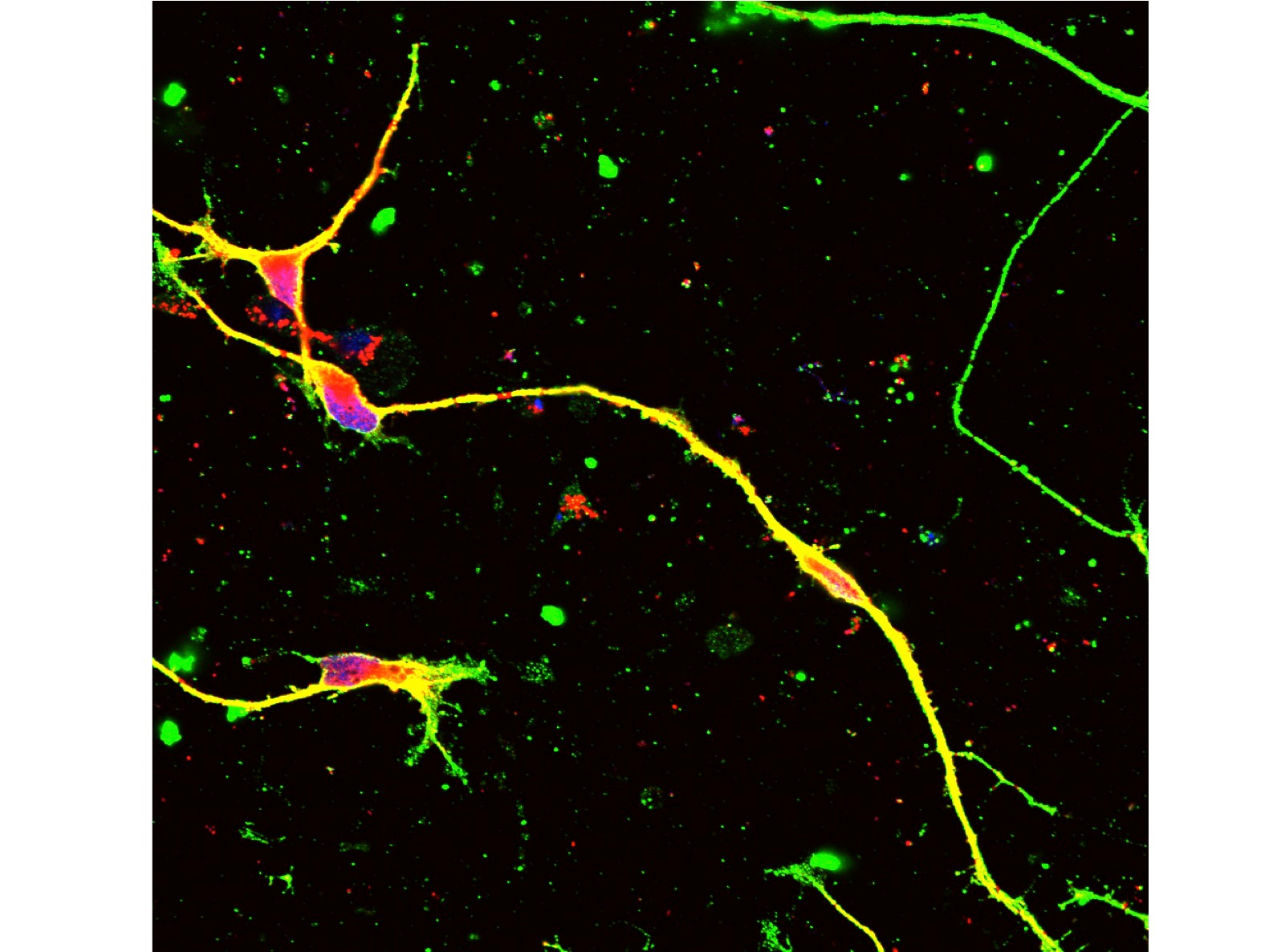
Therefore, we next searched for animal models by which we could test our new method. Unfortunately, transgenic mouse models were not suitable for our examination because transgenic cassettes are too short7,8. Even YAC-transgenic mice were not sufficient as it contains only a part of a single 1.5Mb genome region (ref 9). Hence, we decided to use human CMT1A iPSCs and to develop a Schwann cell-neuron co-culture system. It took years but we could somehow developed the necessary system for examination of our hypothesis. Considering about clinical application in the future, we put the guide RNA with scaffold sequences and human SaCas9 in a single AAV2 vector (Fig 1) and confirmed that at an unexpectedly high efficiency our new method cleaved out an unnecessary 1.5 Mb genome region (Fig 2).

In the Schwann cell-neuron co-culture system, CMT1A mutation increased cell death and hypomyelinated neurites6. Infection of our AAV vector to iPSCs or iPSC-derived Schwann cells recovered both of cell death and hypomyelination by CMT1A mutation in the co-culture6. Moreover, the off-target effect was very rare in the co-culture and in in vivo delivery to normal mice6. Taken together, we concluded that our genome-editing gene therapy for CMT1A could be developed to the next stage. Our strategy is distinct from the other knock-down approaches10-16. After toxicity and pharmacodynamics tests in animals, the next stage will be human clinical trials.

References
- Pearson, T.S. et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat Commun. 11, 4251 (2021).
- Tabrizi, S.J. et al, Phase 1–2a IONIS-HTTRx Study Site Teams. Targeting Huntingtin Expression in Patients with Huntington's Disease. New Engl J Med. 380, 2307-2316 (2019).
- Gutmann, L. & Shy, M. Update on Charcot–Marie–Tooth disease. Curr Opin Neurol 28, 462–467 (2015).
- Li, J., Parker, B., Martyn, C., Natarajan, C. & Guo, J. The PMP22 gene and its related diseases. Mol Neurobiol 47, 673–698 (2013).
- van Paassen, B. W. et al. PMP22 related neuropathies: Charcot-Marie-Tooth disease type 1A and Hereditary Neuropathy with liability to Pressure Palsies. Orphanet J Rare Dis 9, 38 (2014).
- Yoshioka, Y. et al. AAV-mediated editing of PMP22 rescues Charcot-Marie-Tooth disease type 1A features in patient-derived iPS Schwann cells. Commun Med (Lond). 2023 Nov 28;3(1):170.
- Sereda, M. et al. A Transgenic Rat Model of Charcot-Marie-Tooth Disease. Neuron 16, 1049–1060 (1996).
- Magyar, J. P. et al. Impaired Differentiation of Schwann Cells in Transgenic Mice with Increased PMP22 Gene Dosage. J Neurosci 16, 5351–5360 (1996).
- Huxley, C. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet 5, 563–569 (1996).
- Zhao, H. T. et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J Clin Invest 128, 359–368 (2017).
- Lee, J.-S. et al. Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol Dis 100, 99–107 (2017).
- Boutary, S. et al. Squalenoyl siRNA PMP22 nanoparticles are effective in treating mouse models of Charcot-Marie-Tooth disease type 1 A. Commun Biol 4, 317 (2021).
- Gautier, B. et al. AAV2/9-mediated silencing of PMP22 prevents the development of pathological features in a rat model of Charcot-Marie-Tooth disease 1 A. Nat Commun 12, 984–985 (2021).
- Stavrou, M. et al. A translatable RNAi-driven gene therapy silences PMP22/Pmp22 genes and improves neuropathy in CMT1A mice. J Clin Invest 132, e159814 (2022).
- Bolino, A. & D’Antonio, M. Recent advances in the treatment of Charcot‐Marie‐Tooth neuropathies. J Peripher Nerv Syst 28, 134–149 (2023).
- Stavrou, M., Sargiannidou, I., Georgiou, E., Kagiava, A. & Kleopa, K. A. Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies. Int J Mol Sci 22, 6048 (2021).
Follow the Topic
-
Communications Medicine
A selective open access journal from Nature Portfolio publishing high-quality research, reviews and commentary across all clinical, translational, and public health research fields.

Related Collections
With Collections, you can get published faster and increase your visibility.
Public health and health governance in China
Publishing Model: Open Access
Deadline: Jul 31, 2026
Targeted diagnostics and therapies for autoimmune diseases
Publishing Model: Hybrid
Deadline: Oct 31, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in