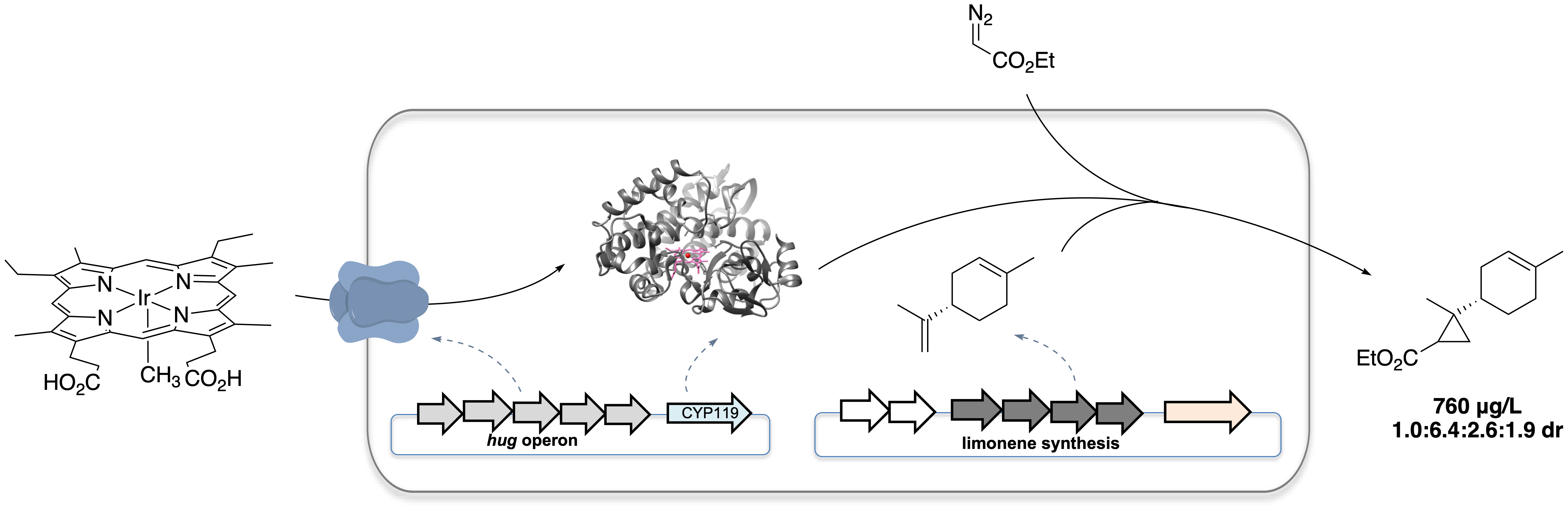
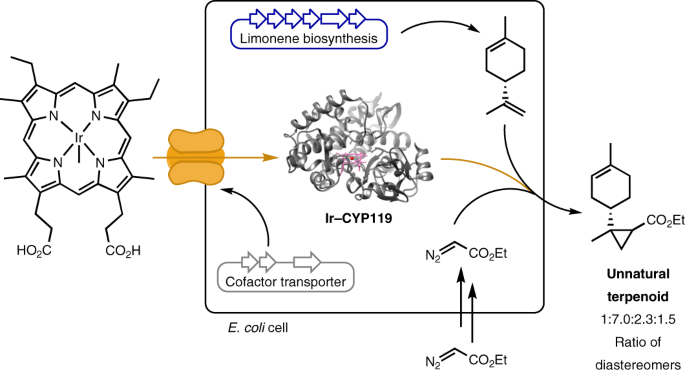
Microbes make organic molecules with complex structures. The rapidly advancing techniques and tools in the field of synthetic biology and metabolic engineering have enabled researchers to design tailor-made biosynthetic pathways and produce non-canonical compounds by reorganizing enzymes from different organisms into a new biosynthetic pathway. The ensuing participation of organic chemists also has expanded the toolbox to access products of non-natural origin by intersecting biosynthetic pathways with small-molecule catalysts or engineering non-natural reactivity in enzymes.
In the past decades, artificial metalloenzymes (ArMs) have emerged as a new class of biohybrid catalysts that merge both the advantages of enzymes and transition-metal catalysts. By affixing transition-metal complexes into a properly engineered protein host, ArMs have been created to catalyze a wide range of reactions, including group transfers to olefins and C-H bonds, cross coupling, asymmetric hydrogenation, and ring-closing olefin metathesis. More recently, a transition towards in vivo application of ArMs has been observed, and the longstanding goal of expanding natural metabolism with in vivo implementation of artificial metalloenzymes seems within reach.
By replacing the native iron in a natural hemoprotein with an abiotic metal, our group has demonstrated that metal substitution creates ArMs that can be evolved to generate enzymes that catalyze abiological reactions with levels of activity and selectivity that can reach those of natural enzymes and on the types of molecules present in natural products that can be produced heterologously in E. Coli. More specifically, the iridium-containing analog of a thermophilic cytochrome P450 (CYP119) from Sulfolobus solfataricus was shown to catalyze the cyclopropanation of unconjugated, hindered terminal alkenes that are present in terpene natural products. This reaction is one of the few in which ArMs catalyze reactions on the types of molecules that could be produced biosynthetically and which demonstrate the potential to combine biosynthesis with catalysis by ArMs.
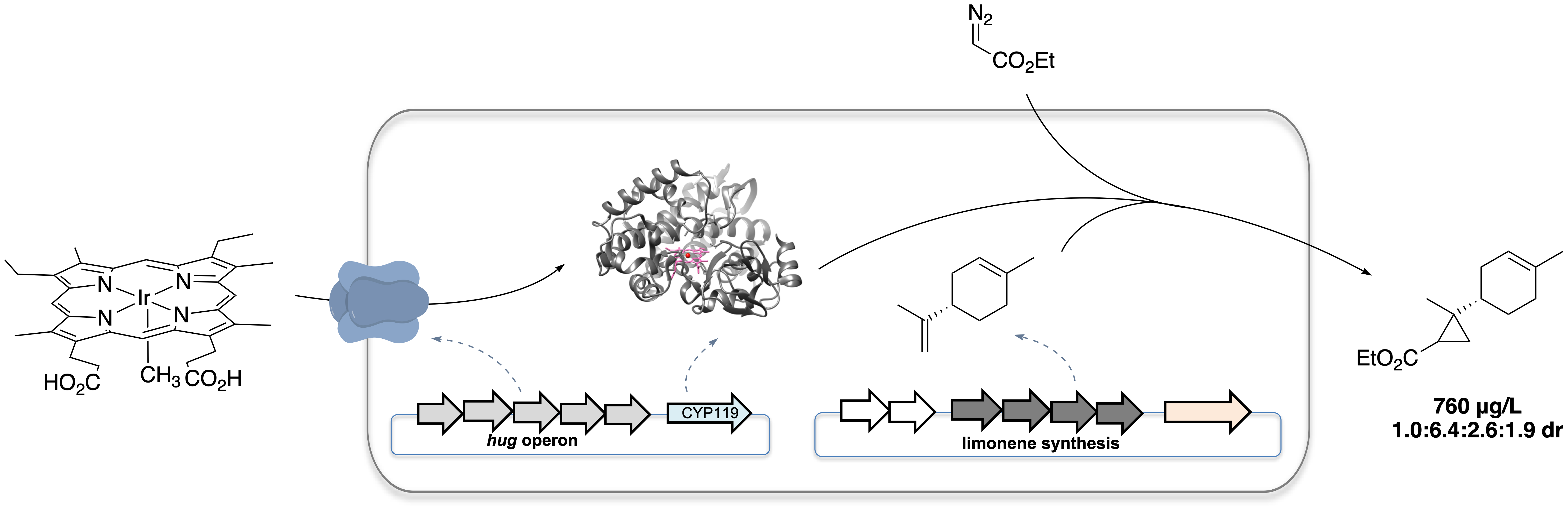
Figure 1. Microbial production of an unnatural terpenoid from an artificial pathway combining natural pathway enzymes and an artificial cytochrome P450.
To accomplish the goal of producing an unnatural terpenoid by incorporating iridium-containing CYP119 (Ir-CYP119) into a terpene biosynthetic pathway, it is necessary first to assemble the artificial metalloenzyme and to conduct reactions of these catalysts in whole cells. To do so, we exploited an outer-membrane heme receptor identified from E. coli O157:H7, ChuA, which has been reported to enable the uptake of heme derivatives bearing modifications on the metal center or porphyrin scaffold into common E. coli lab strains. By over-expressing the heme transporter ChuA and a CYP119 mutant (C317G, T213G, V254L and L155W) in E. coli BL21(DE3) in a culture medium supplemented with 0.02 μM Ir(Me)MPIX (MPIX is mesoporphyrin IX), the whole-cell catalysts harboring Ir-CYP119 assembled in vivo catalyzed the cyclopropanation of (-)-carvone with EDA (ethyl diazoacetate), producing the cyclopropane product with a diastereomeric ratio (d.r., 23:3.5:1.0:1.2) that is much higher than that of products afforded by the same reaction catalyzed by Ir-CYP119 reconstituted from purified apo-CYP119 mutant.
However, incorporation of Ir-CYP119 into a cellular biosynthetic pathway was not straightforward. We found that a balanced expression of enzymes in the terpene pathway and CYP119 was essential to obtain the desired product in substantial amounts. Expressing a limonene biosynthetic pathway heterologously in E. coli could produce limonene with a high titer of 188 mg/L, while co-expressing the pathway with CYP119 regulated by a T7 promoter caused the titer to decrease drastically to 28 mg/L. We therefore needed to express CYP119 with a weaker promoter, such as the lacUV5 promoter, to restore the production of limonene. However, when we attempted to conduct the reaction of (−)-carvone with EDA with cells co-expressing ChuA and CYP119 with the lacUV5 promoter, the cyclopropane was formed with low diastereoselectivity, implying that assembly of the ArM was inefficient under this condition. Thus, efficient incorporation of iridium porphyrin needed to occur with low expression levels of CYP119. Fortunately, an alternative heme transport system, HUG, which expresses the complete Plesiomonas shigelloides heme utilization gene operon, was found to efficiently transport the artificial cofactor and allowed the whole-cell cyclopropanation of (−)-carvone to occur with a high d.r. of 24.0:3.1:1.0:1.2 even with a low expression level of CYP119.
The next step was to integrate such diastereoselective cyclopropanation with the biosynthesis of terpenes. Although (-)-carvone has been produced from glucose in E. coli, the presence of intermediates with structures similar to each other along the pathway and the low titer of the carvone produced biosynthetically would complicate analysis of the unnatural terpenoid products. Thus, we chose to combine the production of (-)-limonene with the reactivity of Ir-CYP119 because it is a structurally related monoterpene known to be produced in high titer in E. coli. Indeed, the whole-cell cyclopropanation with limonene added exogeneously catalyzed by E. coli cells co-expressing the HUG system and CYP119 with lacUV5 promoter produced the cyclopropyl limonene with a substantial d.r. (1.0 : 3.3 : 1.1 : 1.0), which is different from that of the reaction catalyzed by free Ir(Me)MPIX (1.0 : 1.2 : 3.5 : 4.2 dr). After combining limonene production and artificial catalysis within the same cell, the artificial pathway produced 250 μg/L of cyclopropyl limonene with a d.r. of 1.0:3.2:1.8:1.5. The titer and diastereoselectivity of the cyclopropane product was further increased by evolving a more selective CYP119 mutant and optimizing the feeding rate of EDA. With the incorporation of this new mutant into the pathway and batch-wise additions of EDA, the unnatural terpenoid was produced with a higher titer of 760 μg/L, and a higher d.r. of 1.0:6.4:2.6:1.9.
By combining the capabilities of synthetic biology to produce natural and unnatural products in heterologous hosts with the wide range of abiotic reactions of artificial metalloenzymes from chemical catalysts in protein hosts, one can envision that the alliance between natural biosynthesis and artificial metalloenzyme will, in the near future, create microorganisms that produce a wide range of unnatural products that contain new functional groups on the periphery of complex architectures, new core structures generated by chemists’ ability to conduct cyclization and insertion reactions, and new core structures generated by synthetic biologists’ ability to manipulate nature’s biosynthetic pathways.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in