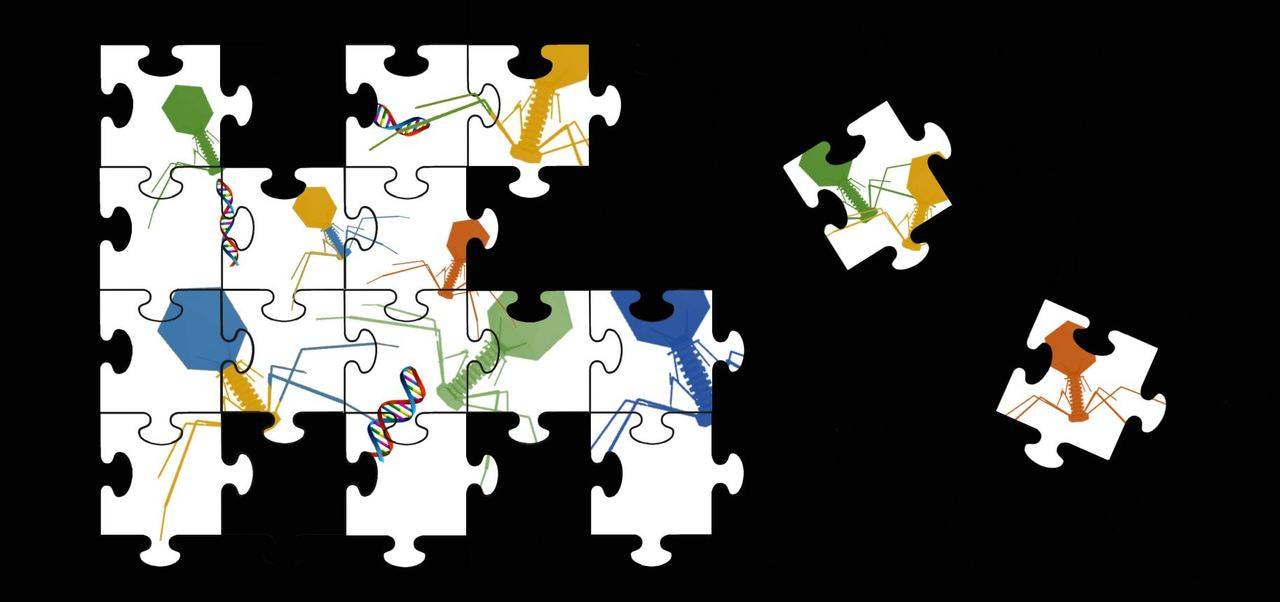

The human gut virome consists primarily of bacteriophage which may both play a crucial role in regulating and shaping microbial communities of the gut and facilitate horizontal gene transfer and microbial evolution. With 90% of viral sequencing reads sharing little to no homology to reference databases, the make-up of these viral communities also represents one of the biggest gaps in our understanding of the human microbiome. As the hosts of the majority of these viruses are also unknown, the virome research community relies heavily on sequencing and computational approaches.
When we developed our virome analysis pipeline we realised that if we were to make sense of this unknown majority we would need to move towards In silico methods and away from database dependent approaches. Our first port of call was a crucial stage in all reference-independent pipelines, the assembly step, at which short sequence reads are used to recreate the genome sequences of community members. Looking at previous studies we realised that there was no single assembly method used across all virome studies, nor had there been an extensive assembly comparison dedicated to the virome, which led us to this study.
Metagenomic assembly, or reconstructing the genome sequences of community members, is a common but challenging computational task due to the complexity of microbial communities and large amounts of sequencing data required to represent them in a meaningful way. Unfortunately for virome scientists, assembly challenges of viromes are more difficult – perhaps even by orders of magnitude. The ability of the assembler to overcome these challenges is of significant importance to a virome analysis pipeline, which is essentially built around this crucial step.
By testing 16 assembly approaches on a combination of 4 different virome datasets including both synthetic and human viromes, we observed significant variation in assemblers’ ability to overcome assembly challenges. Most assemblers failed to properly reconstruct phage genomes that we knew to be there, which was a worrying outcome. In most cases the assemblers resulted in only small proportions of the genomes being recovered and assemblies being short and fragmented. These findings have serious implications for virome analysis pipelines, as not only does the choice of assembly program used in a study directly impact which members of a viral community can be recovered, but certain viral genomes appear to challenge all current assembly approaches. We observed that extremes in abundance were responsible for aspects of poor assembly, as were the proportion of genomic repeat regions in each community member. However, these challenges did not explain the full variation in poor genome recovery, highlighting a continued need to improve and develop virome analysis approaches as well was important considerations when setting downstream analysis parameters and making final conclusions.
Follow the Topic
-
Microbiome

This journal hopes to integrate researchers with common scientific objectives across a broad cross-section of sub-disciplines within microbial ecology. It covers studies of microbiomes colonizing humans, animals, plants or the environment, both built and natural or manipulated, as in agriculture.
Related Collections
With Collections, you can get published faster and increase your visibility.
Harnessing plant microbiomes to improve performance and mechanistic understanding
This is a Cross-Journal Collection with Microbiome, Environmental Microbiome, npj Science of Plants, and npj Biofilms and Microbiomes. Please click here to see the collection page for npj Science of Plants and npj Biofilms and Microbiomes.
Modern agriculture needs to sustainably increase crop productivity while preserving ecosystem health. As soil degradation, climate variability, and diminishing input efficiency continue to threaten agricultural outputs, there is a pressing need to enhance plant performance through ecologically-sound strategies. In this context, plant-associated microbiomes represent a powerful, yet underexploited, resource to improve plant vigor, nutrient acquisition, stress resilience, and overall productivity.
The plant microbiome—comprising bacteria, fungi, and other microorganisms inhabiting the rhizosphere, endosphere, and phyllosphere—plays a fundamental role in shaping plant physiology and development. Increasing evidence demonstrates that beneficial microbes mediate key processes such as nutrient solubilization and uptake, hormonal regulation, photosynthetic efficiency, and systemic resistance to (a)biotic stresses. However, to fully harness these capabilities, a mechanistic understanding of the molecular dialogues and functional traits underpinning plant-microbe interactions is essential.
Recent advances in multi-omics technologies, synthetic biology, and high-throughput functional screening have accelerated our ability to dissect these interactions at molecular, cellular, and system levels. Yet, significant challenges remain in translating these mechanistic insights into robust microbiome-based applications for agriculture. Core knowledge gaps include identifying microbial functions that are conserved across environments and hosts, understanding the signaling networks and metabolic exchanges between partners, and predicting microbiome assembly and stability under field conditions.
This Research Topic welcomes Original Research, Reviews, Perspectives, and Meta-analyses that delve into the functional and mechanistic basis of plant-microbiome interactions. We are particularly interested in contributions that integrate molecular microbiology, systems biology, plant physiology, and computational modeling to unravel the mechanisms by which microbial communities enhance plant performance and/or mechanisms employed by plant hosts to assemble beneficial microbiomes. Studies ranging from controlled experimental systems to applied field trials are encouraged, especially those aiming to bridge the gap between fundamental understanding and translational outcomes such as microbial consortia, engineered strains, or microbiome-informed management practices.
Ultimately, this collection aims to advance our ability to rationally design and apply microbiome-based strategies by deepening our mechanistic insight into how plants select beneficial microbiomes and in turn how microbes shape plant health and productivity.
This collection is open for submissions from all authors on the condition that the manuscript falls within both the scope of the collection and the journal it is submitted to.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Microbiome, Environmental Microbiome). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Collection policies for Microbiome and Environmental Microbiome:
Please refer to this page. Please only submit to one journal, but note authors have the option to transfer to another participating journal following the editors’ recommendation.
Collection policies for npj Science of Plants and npj Biofilms and Microbiomes:
Please refer to npj's Collection policies page for full details.
Publishing Model: Open Access
Deadline: Jun 01, 2026
Microbiome and Reproductive Health
Microbiome is calling for submissions to our Collection on Microbiome and Reproductive Health.
Our understanding of the intricate relationship between the microbiome and reproductive health holds profound translational implications for fertility, pregnancy, and reproductive disorders. To truly advance this field, it is essential to move beyond descriptive and associative studies and focus on mechanistic research that uncovers the functional underpinnings of the host–microbiome interface. Such studies can reveal how microbial communities influence reproductive physiology, including hormonal regulation, immune responses, and overall reproductive health.
Recent advances have highlighted the role of specific bacterial populations in both male and female fertility, as well as their impact on pregnancy outcomes. For example, the vaginal microbiome has been linked to preterm birth, while emerging evidence suggests that gut microbiota may modulate reproductive hormone levels. These insights underscore the need for research that explores how and why these microbial influences occur.
Looking ahead, the potential for breakthroughs is immense. Mechanistic studies have the power to drive the development of microbiome-based therapies that address infertility, improve pregnancy outcomes, and reduce the risk of reproductive diseases. Incorporating microbiome analysis into reproductive health assessments could transform clinical practice and, by deepening our understanding of host–microbiome mechanisms, lay the groundwork for personalized medicine in gynecology and obstetrics.
We invite researchers to contribute to this Special Collection on Microbiome and Reproductive Health. Submissions should emphasize functional and mechanistic insights into the host–microbiome relationship. Topics of interest include, but are not limited to:
- Microbiome and infertility
- Vaginal microbiome and pregnancy outcomes
- Gut microbiota and reproductive hormones
- Microbial influences on menstrual health
- Live biotherapeutics and reproductive health interventions
- Microbiome alterations as drivers of reproductive disorders
- Environmental factors shaping the microbiome
- Intergenerational microbiome transmission
This Collection supports and amplifies research related to SDG 3, Good Health and Well-Being.
All submissions in this collection undergo the journal’s standard peer review process. As an open access publication, this journal levies an article processing fee (details here). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief.
Publishing Model: Open Access
Deadline: Jun 16, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in