Chemicals from CO2 + green electricity: The electrified synthesis of ethylene oxide
Published in Chemistry
Ethylene oxide (EO) is a commodity chemical produced at 20 million tons/annum. It is used in the manufacture of polyethylene terephthalate (PET), and it also makes its way into fabrics like spandex. It also has direct uses of its own, serving as a disinfectant in hospitals.
Since it is such a major chemical product, and today it is derived from fossil ethylene, it of interest to determine how we can instead consume CO2 in EO’s production. Existing processes for EO production release ~2 tCO2/tEO (Ref. 1). Electrochemical processes that produce EO from CO2 – when powered using renewable energy – enable a net reduction in CO2 emissions: indeed they consume 2 tCO2/tEO.
An electrochemical process was recently reported that produces EO from CO2 using two independent electrolyzers: first was CO2 reduction to ethylene (C2H4), and this was then oxidized to EO (Ref. 2). Unfortunately, this literature benchmark system suffered from a limited CO2-to-EO faradaic efficiency (FE), a value of 6%. As a result, the energy required was ~19 MJ per kg of EO, ~5x more energy-intensive than today’s thermochemical route (Ref. 3).
We found that this arose due to the presence of hypochlorous acid (HOCl) cleavage to unreactive ClO− (Fig. 1) – and thus that if we could suppress HOCl cleavage, we could improve the C2H4-to-EO FE and enhance the conversion of CO2-to-EO.
We explored therefore barium, lanthanum, cerium, and bismuth oxides as candidates for suppressing HOCl cleavage. Of these catalysts, the barium oxide loaded iridium oxide (BaOx/IrO2) showed the best results: it limited the FE towards unreactive ClO− to below 10%, thus increasing the C2H4-to-EO FE to 90%. DFT results showed that HOCl cleavage should no longer occur.
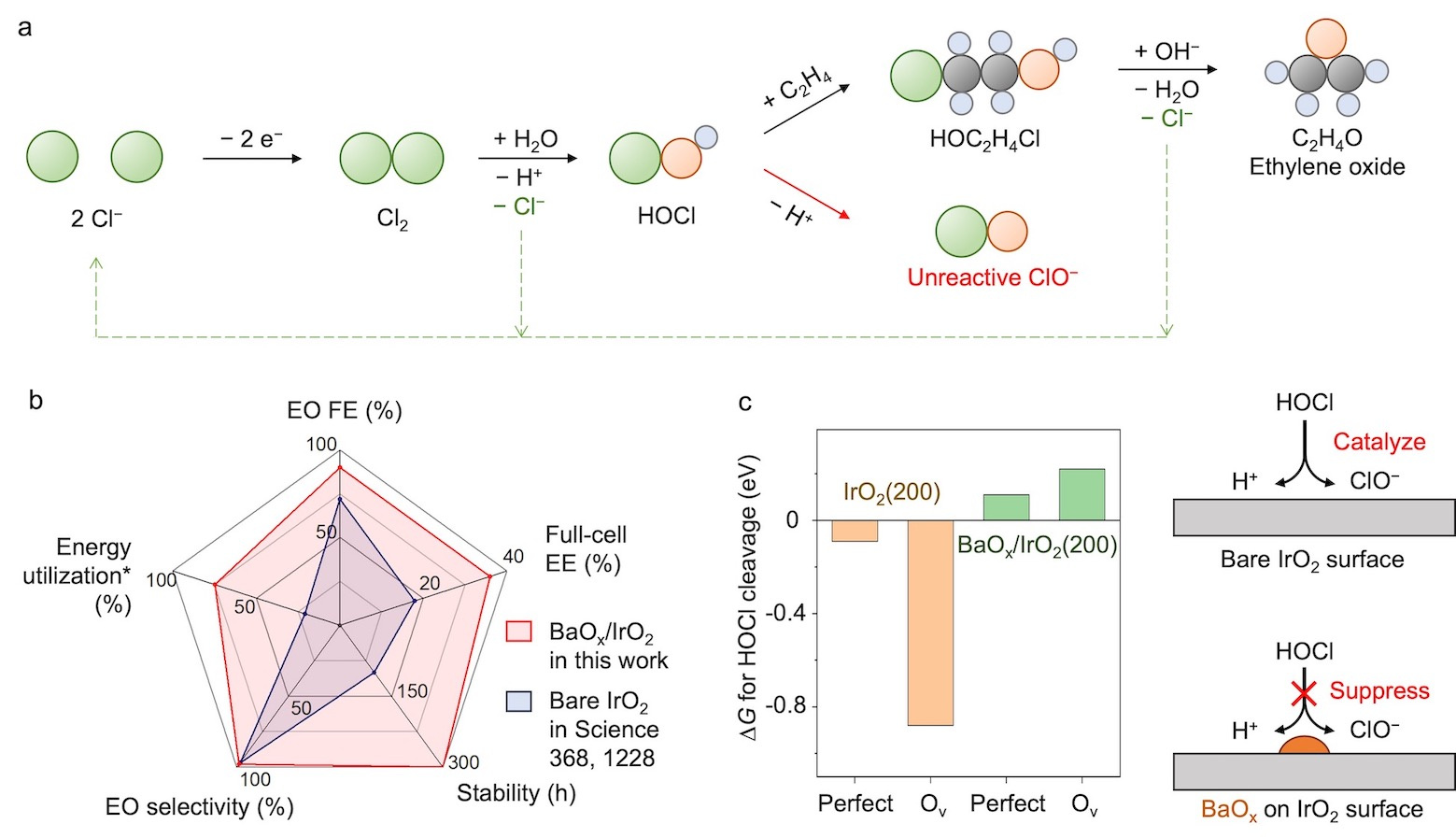
Fig. 1 | (a) Reaction pathway to produce EO using HOCl as the key intermediate. (b) Comparison of EO FE, full-cell EE, stability, EO selectivity, and energy utilization compared to best prior results for EO electrosynthesis. (c) Gibbs free energy changes (∆G) of the reaction *HOCl → *H + *OCl on bare IrO2(200) and Ba3O4-cluster loaded IrO2(200) with perfect and oxygen-vacancy (Ov) surfaces.
However, the high voltage remained a problem: the energy input was still impractical, ~9 MJ/kg of EO, still 2.2x higher than in existing thermochemical processes. We noted that one significant factor was that – in addition to converting C2H4 to EO on the anodic (oxidative) side of the reactor, we were also producing hydrogen on the cathode, an energy-intensive process.
We believed that if we could displace HER at the cathode and replace it with a lower-thermodynamic-potential reaction, we would significantly reduce the energy input of overall C2H4-to-EO conversion.
We replaced HER with the oxygen reduction reaction (ORR), and found that we maintained >80% FE for C2H4-to-EO and reduced the operating full-cell voltage by fully 1.2 V. We lowered the electrical energy input to 5.3 MJ/kg of EO, close to that of the conventional (emissions-intensive) industrial process for producing EO (~4 MJ/kg of EO).
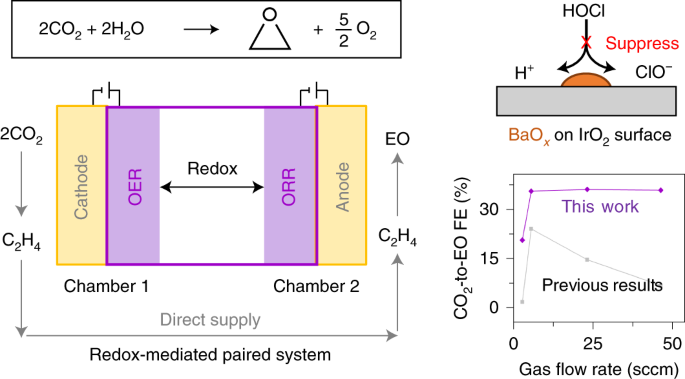
We then built a full prototype system, an oxygen redox (H2O/O2) mediated paired system that produced EO from CO2 (Fig. 2). We paired up CO2-to-C2H4 reduction in a first stage of the cascade system with C2H4-to-EO oxidation the second stage, connecting the two using a H2O/O2 mediator that cycles between the oxygen evolution reaction (OER) and ORR.
By using this system, we also overcame the larger electron consumption in CO2-to-C2H4 (12 e−) vs. C2H4-to-EO (2 e−) by converting more H2O into O2, rendering H2O the only sacrificial agent.
As a result, by using the redox mediated paired system, we achieved a 1.5-fold higher CO2-to-EO FE (35%) and used a 1.2 V lower operating voltage than literature benchmark electrochemical systems (Ref. 2).
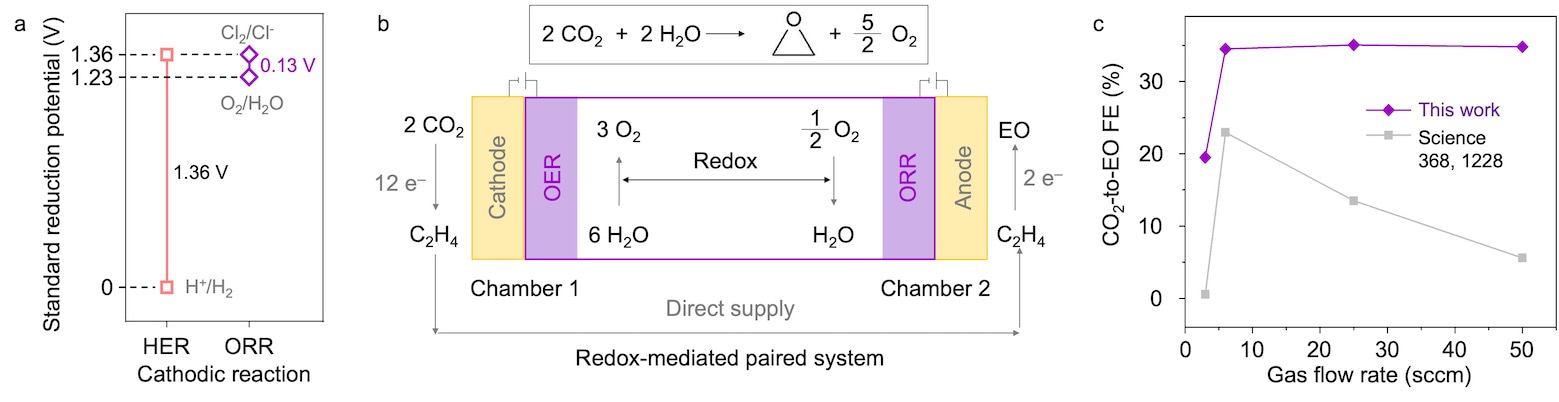
Fig. 2 | (a) Theoretical reaction potentials for producing EO when the cathode is HER or ORR. (b) Total reaction equation and schematic description of the electrochemical process to produce EO from CO2. (c) Comparison of CO2-to-EO FEs using BaOx/IrO2 catalysts herein relative to that in prior reports (Ref. 2).
For more details, please check out our paper “Redox-mediated electrosynthesis of ethylene oxide from CO2 and water” in Nature Catalysis.
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in