Comparison and harmonization of targeted bisulfite-sequencing platforms
Published in Bioengineering & Biotechnology
Epigenetics invokes upregulation or downregulation of gene expression without changing the underlying DNA sequence. Regulation of gene expression by DNA methylation is crucial for the survival and adaptability of living organisms as a response to developmental cues and environmental stimuli.
Most of the DNA methylation-based research in epigenomics over the past two decades was based on microarray technology. In recent years, the development of targeted bisulfite sequencing (TBS) methods has made it possible to comprehensively analyze the human methylome. Both microarray methods and NGS are based on single-base resolution sequencing of bisulfite-converted DNA. While microarrays are certainly more affordable than NGS, with a lower computational footprint, NGS has a big advantage in that it can read single molecules and allows CpG phasing to define epiallele status and study intra-sample heterogeneity.
In 2015 several exciting studies came out looking at the intra-tumoral epigenetic heterogeneity in cancer. These studies used in-house protocols for reduced-representation bisulfite sequencing (RRBS). At the Medical Genomics Group at UCL Cancer Institute led by Prof. Stephan Beck, we also wanted to study tumor heterogeneity in several solid tumors, but for large clinical cohorts, it is important to minimize technical variation, so a commercial kit with standardized reagents and protocol is preferred.

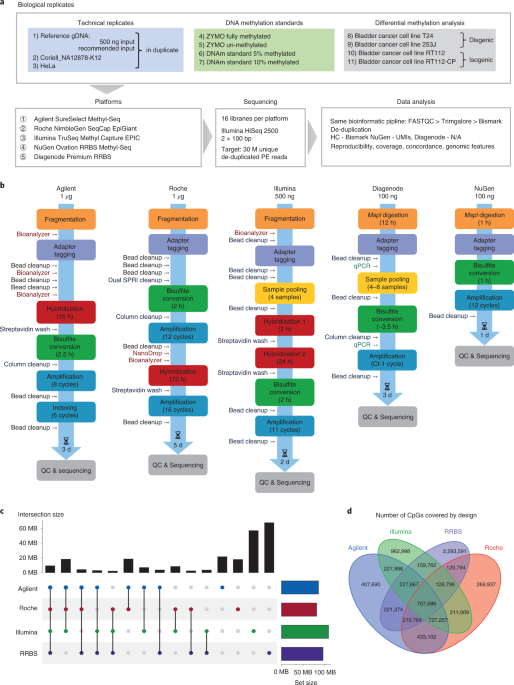
On the market, several companies were offering standardized off-the-shelf kits for TBS based on hybridization capture from Agilent, Roche, and Illumina, and two - Diagenode and NuGen, offering standardized RRBS library preparation kits.
But which platform to choose? While trying to decide which one to use, as there were no guidelines or comparisons between the platforms, we decided to compare them ourselves.
To mimic a real-life experimental setup, we selected 11 different samples, including well-characterized cell lines such as Coriell NA12878 and HeLa, a pool of blood samples representing a heterogeneous mix of cell types, commercial DNA methylation standards, and two pairs of cell lines with different phenotypes to compare differential methylation calls - one pair with dysgenic and second with isogenic origin. Using the same DNA from these 11 samples, we evaluated the sequencing cost-effectiveness, reproducibility, and concordance between the TBS platforms, and compared the results with those obtained by a ‘gold standard’ technique based on traditional bisulfite-based whole-genome sequencing and the third-generation Nanopore technology.
All of the kits performed well and generated robust and reproducible data, but with different sequencing cost-effectiveness. The study also highlighted the importance of using molecular barcoding to overcome technical biases in DNA methylation calling. However, it proved difficult to compare the results generated by the different kits, partly because they each target a somewhat different set of CpGs in the human genome.
For example, as some platforms focus more on the promoter regions, while others on enhancers this study will help guide researchers working in different fields towards the most appropriate method for their specific application. The study also makes an important contribution to the FAIR Data Principles (Findable, Accessible, Interoperable, and Reusable) by improving the interoperability and reusability of data generated on different commercial platforms. To achieve this, we proposed a framework to integrate the data generated using different platforms, either by focusing on differentially methylated regions rather than individual CpGs, or by harmonizing the datasets using a computational method to impute missing CpG sites.
This work will hopefully make life easier for many scientists banging their heads with Buridán's donkey question – which TBS platform to choose? Finally, it will help researchers compare ‘apples and oranges’ and increase the power of their studies by integrating datasets generated using different platforms or reproducing their findings.
I am a molecular biologist with a PhD in human cancer genetics. My primary research interests lie in the field of cancer genetics and epigenetics, applying high-throughput genomics and bioinformatics methods, complemented with in vitro functional studies.
My research aims for a better understanding of cancer etiology and improved patient management through the development of diagnostic, prognostic, and predictive biomarkers.
My recent work focuses on the (epi)genetic epidemiology of solid tumors and the development and application of novel technologies for the detection of (epi)genetic variants from liquid biopsies.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in