Decoding translation in pathogenic Salmonella and Listeria
Published in Microbiology and Cell & Molecular Biology

Infectious diseases stand as the leading cause of death worldwide. Many diverse bacterial pathogens employ common mechanisms to adhere to, invade, and establish infections within host cells. Non-typhoidal serovars of Salmonella, the leading cause of food-borne gastroenteritis, infect millions and result in approximately 230,000 fatalities each year. The gram-positive bacterium Listeria monocytogenes, the primary cause of listeriosis, ranks among the most virulent food-borne pathogens, with a high mortality rate associated with infection. Both Salmonella and Listeria encode a range of virulence factors to promote entry and survival within host cells. Key examples include large macromolecular secretion systems that release effector proteins into host cells to facilitate invasion, and adhesins that assemble on the bacterial cell surface to enhance attachment to host cells or other surfaces. These virulence factors are often encoded by genes located in large clusters containing multiple operons.
Significant progress has been made in understanding bacterial transcription. The development of deep sequencing and RNA-seq has allowed the study of transcription at the nucleotide level1,2. However, despite the close linkage between prokaryotic transcription and translation, transcript levels often do not correlate with protein synthesis, meaning that RNA-seq alone is not sufficient to understand gene expression3.
Through paired high-definition translatomic and transcriptomic studies, we can, for the first time, visualize and accurately quantify translation efficiency for all translated mRNAs in these two highly divergent pathogenic bacteria. We have confirmed that translational control plays a major role in controlling the stoichiometry of components for many virulence machineries, including the SPI-1 injectisome. Furthermore, in Salmonella, efficiently translated mRNAs generally have either a strong Shine-Dalgarno (SD) sequence or weak secondary structure surrounding the SD site, aligning with the long-established model for efficient translation initiation observed in other closely related Gram-negative bacteria like E. coli. Additionally, we identified a novel cis-regulatory feature where the presence of hexa-adenine next to the start codon drives efficient translation in both Salmonella and E. coli. Strikingly, none of these features (strong SD sequence, weak secondary structure surrounding the SD sequence, or hexa-adenine) are associated with efficient translation in Listeria, a highly divergent Gram-positive bacterium from the Firmicutes phylum.
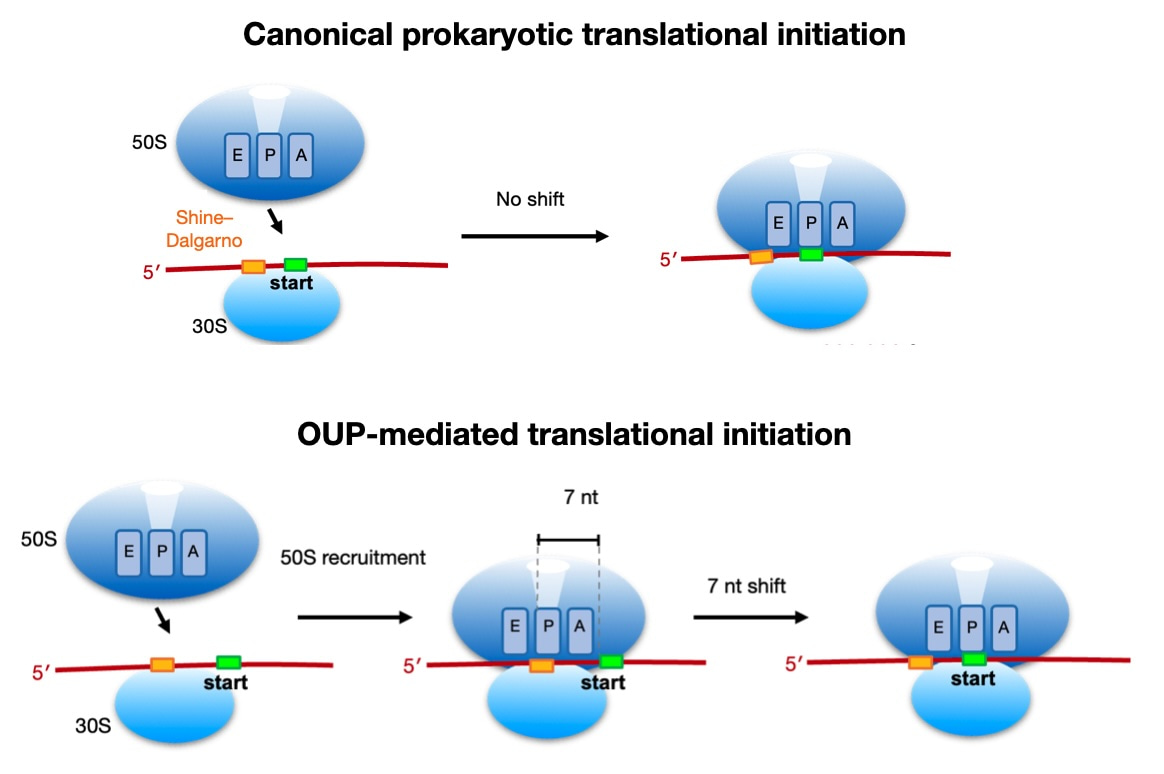
Finally, we identified a potentially novel initiation mechanism in Listeria, utilized by 15% of genes, where the formation of the 70S ribosome occurs 7 nt upstream of the canonical assembly location (i.e., with the start codon positioned at the P-site). We term this potentially novel mechanism 'Out-of-frame Upstream Peak' (OUP)-mediated initiation. Despite comprising only 16% of genes, OUP genes tend to be highly transcribed and efficiently translated, accounting for approximately 40% of all proteins produced in Listeria. Currently, we are investigating the detailed molecular mechanisms involved in OUP-mediated translation initiation.
Our work highlights the striking differences in translational regulation between these two evolutionarily divergent pathogenic bacteria, thus advancing our understanding of translation control in these major human pathogens.
References
- Sultan, M. et al. A Global View of Gene Activity and Alternative Splicing by Deep Sequencing of the Human Transcriptome. Science (1979) 321, 956–960 (2008).
- Wilhelm, B. T. et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453, 1239–1243 (2008).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S. & Weissman, J. S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science (1979) 324, 218–223 (2009).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Very nice work. Thanks for sharing.