Determining the adsorption energies of small molecules with the intrinsic properties of adsorbates and substrates
Published in Electrical & Electronic Engineering
The adsorption of a molecule to a solid surface is a general phenomenon in many processes, that is, heterogeneous catalysis, gas sensors, molecular electronics, biomedical applications, and so on. It is widely accepted that the adsorption properties are determined by the electronic and geometric structures of substrates and adsorbates. Therefore, identifying the electronic and geometric determinants of adsorption and the underlying mechanism has been a long-term goal in past decades. Despite the great efforts by the well established models and schemes, such as the d-band model, generalized coordination number model and the linear scaling relationships, there still lacks a formula or a relationship to bridge the gap between the adsorption energies and the easily accessible intrinsic properties of adsorption systems, particularly by means of incorporating the electronic and geometric properties.
We provide a far-reaching solution to this issue, by identifying that the three main factors that control the adsorption energies are the valence and electronegativity of surface atoms, the coordination of active sites, and the valence of adsorbates. The expression for transition metals (TMs) and nanoparticles is as follows,
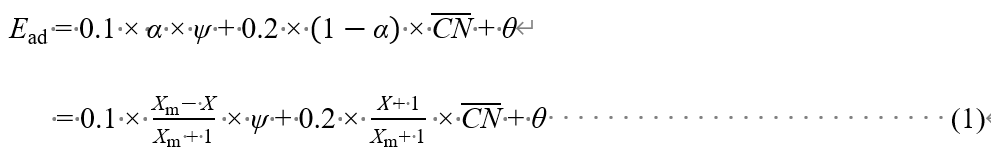
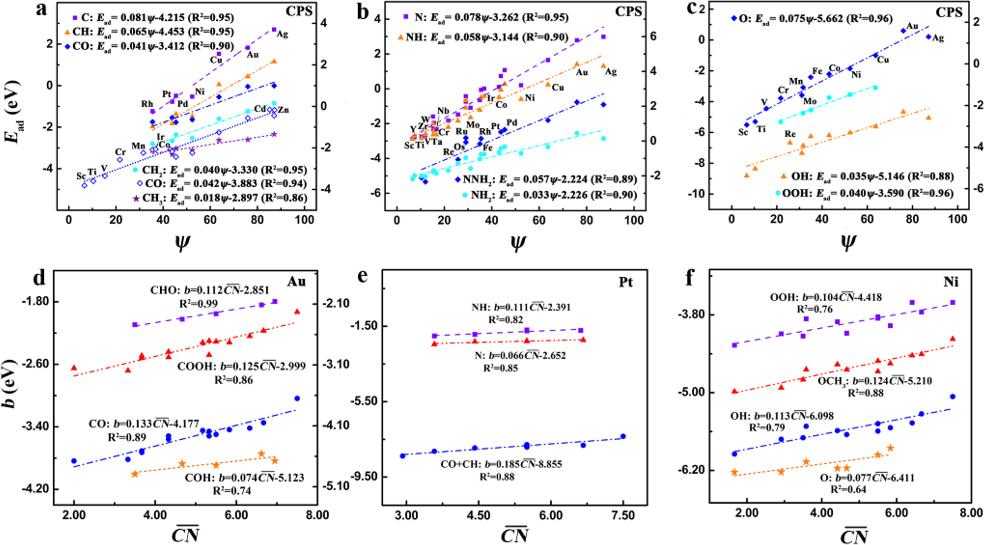
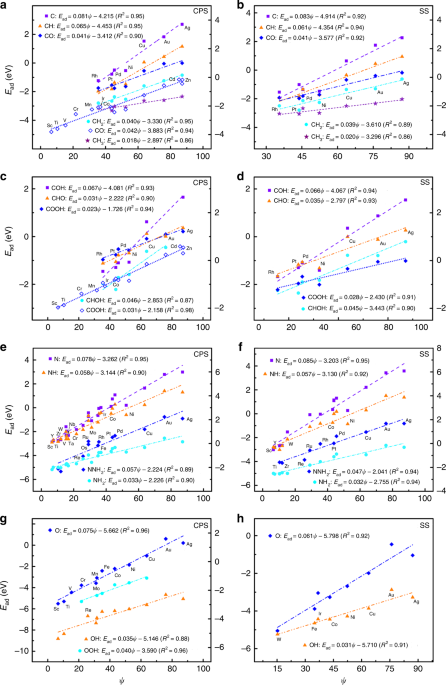
where Xm is the maximum number that the central atom of a given adsorbate can bond to the specific coordinating group, and X is correspondingly the actual number. ψ is a novel electronic descriptor based on the number of valence electrons (Sv) and electronegativity (χ) of TMs for describing adsorption properties as ψ = Sv2/χ. The adsorption energies scale linearly with ψ for the variable materials with the same geometric sites (see Fig. 1a-c) and with the generalized coordination number for the various facets of a given material (see Fig. 1d-f). The constant θ is the only parameter that needs to be determined, e.g. through DFT calculations, while the rest parameters are intrinsic and readily accessible.

One of the key advantages of our scheme is that it can be naturally generalized into near-surface alloys (NSAs) and oxides by including the local environment effects of active centers. This is particularly encouraging, since the adsorption of OH, F, and Cl on NSAs and the adsorption of OHx on oxides cannot be accurately described by the d-band model. Our results demonstrate that the descriptor ψ essentially reflects the upper edge of the d-states for TMs and is linked with the outer-electron characteristics of surface atoms for oxides.
Our model is descriptive and predictive for twenty species on transition metals, nanoparticles, near-surface alloys, and oxides, and can derive automatically the linear scaling relationships of the adsorption energies and its generalized form, reflecting the solid physical-chemical basis. This fully predictive scheme uncovers the fundamental physical rules of adsorption, generalizes the efficiency and limitation of engineering the adsorption energy and reaction energy, and allows rapid screening of potentially interesting systems since all involved parameters are predictable, all of which provide a long-sought guide for future materials design.
For more details, please see our recent publication in Nature Communications: Determining the adsorption energies of small molecules with the intrinsic properties of adsorbates and substrates (https://doi.org/10.1038/s41467-020-14969-8).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in