DIALOGUE maps multicellular programs in tissue from single-cell or spatial transcriptomics data
Published in Bioengineering & Biotechnology


The cell is the fundamental unit of life, yet many biological processes operate at higher functional scales, spanning multiple cell types within and across tissues. Therefore, changes in the tissue structure can be used as diagnostics, and therapeutic strategies like immunotherapies that target cell-cell interactions can be extremely powerful. But mapping regulatory processes across cells has been a challenge. In this study, we present the first data driven method to map expression regulation across cells, delineate the cell state as a function of its environment, and map such multicellular programs in health and disease.
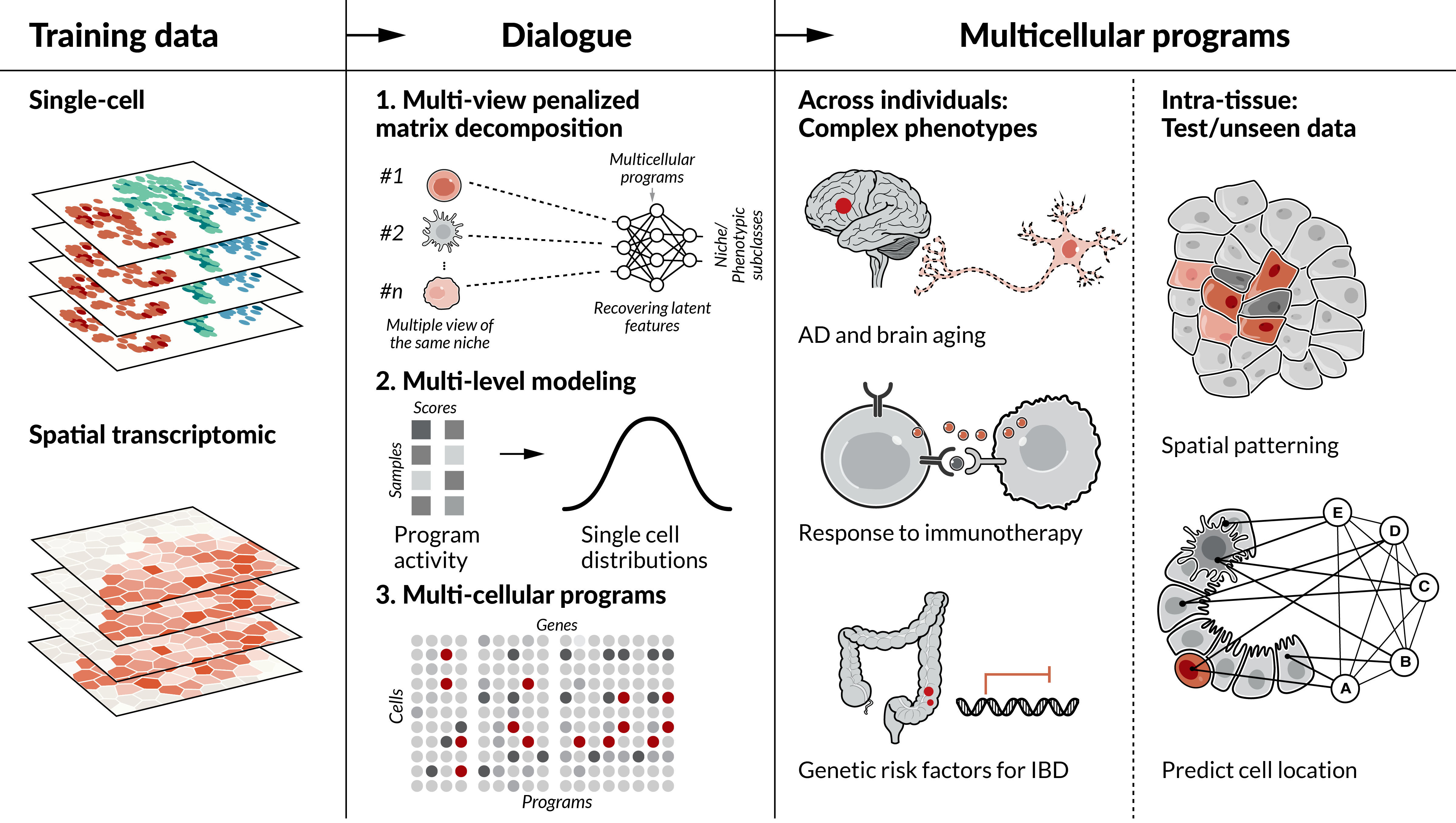
Single cell sequencing and more recent advances in spatial genomics now allow us to profile thousands to millions of cells in their native environment in intact tissues, preserving and recording their physical location. Most studies to date have used these types of data to study cellular states and handled different cells residing in the same tissue as independent entities (including in the standard statistical and computational analysis methods they applied).
However, cells that reside next to or even far away from each other in the same niche, tissue or organism are typically not independent, because they are impacted by shared cues from their micro- or macro-environment, often share genetic/epigenetic information or lineage, and can also directly impact one another.
In this study, we sought to leverage this property for the first time to recover multicellular regulation from single cell and spatial transcriptomics. For this, we first introduced the concept of Multicellular Programs, or MCPs, and then developed the first method to systematically uncover MCPs from single-cell or spatial data.
What are multicellular programs?
Different cell types in one niche or tissue are expected to coordinate their actions: either because they are all responding to a shared signal or because one of the cells responds to a cue and affects the others. This can trigger a collection of different cell types to simultaneously activate the same, cell-type-independent program, or, more typically, different yet highly coordinated cell-type-specific programs. We call this coordinated response of the same or different genes across different cell types a Multicellular Program (MCP).
How can we find Multicellular Programs (MCPs) from data?
DIALOGUE, the method we developed for finding multicellular programs, looks for cross-cell-type co-variation patterns from either spatial or single cell genomics data. Typical analysis of expression programs looks for genes that co-vary within single cells of one type. DIALOGUE looks for (possibly different) genes that are expressed in different cell types, but co-vary in the different cells across niches or samples. In this way it identifies corresponding expression programs across different cell types – where the expression of one set of genes in a certain cell is associated with the expression of the same or another set of genes in nearby cells or cells from the same sample/niche. As a result, a multicellular program found by DIALOGUE reflects a scenario where when cell type A activates one program, cell type B activates (or represses) another program.
How does DIALOGUE work? Given single-cell/spatial data, DIALOGUE treats different cell types from the same micro/macro-environment or sample as different representations of the same entity. It then identifies MCPs in two steps. First, it identifies sparse canonical variates that transform the original feature space to a new feature space, where the different, cell-type-specific, representations are correlated across the different samples/environments. Second, it identifies the specific genes that comprise these latent features by fitting multilevel mixed-effects models that account for single-cell distributions and control for confounders. As output, DIALOGUE provides MCPs, such that each MCP is composed of multiple gene subsets (one per cell type). DIALOGUE is provided as an R package via GitHub.
How did we use our new method and what did we find?
First, we confirmed our hypothesis that different sets of genes across different cell types are coordinated together in the tissue by applying DIALOGUE to different types of spatial transcriptomic datasets, including MERFISH1, Slide-Seq2, and seqFISH3, spatially annotated scRNA-Seq data4 collected across different parts of the mouse brain, and human tumor tissues profiled spatially by SMI5. These coordinated MCPs were highly generalizable: when we used a model trained on a subset of the data, and tested it on unseen data, it correctly predicted the expression of the cell's neighbors based on the cell's own genes expression and the MCPs it learned in the training. We also showed that such signals were not recovered with other existing methods.
And there was another neat application: With the MCPs at hand, we showed that we can predict which micro/macro-environment a cell came from just based on its own transcriptome, and spot cells that are “newcomers” or “unrelated” to the environment.
Notably, some of the strongest programs we found were not “spatial’ per se, and instead showed substantial co-variation across different brain samples. When we examined the non-spatial brain MCPs further, for example, we found that they were associated with mouse behavior and learning experiences.
This prompted us to leverage the same concepts and statistical modeling to identify MCPs based on single cell profiles of dissociated cells collected across different conditions, samples, and individuals, and examine if such MCPs could also mark complex phenotypes in different human disease contexts.
What did we learn about human disease by applying DIALOGUE to scRNA-seq data?
We applied DIALOGUE to scRNA-seq or snRNA-seq from three human diseases: (1) ulcerative colitis, a complex disease that results in colorectal inflammation and ulcers; (2) Alzheimer’s disease; and (3) melanoma.
Ulcerative colitis
One of the most notable MCPs spanned five different cell types, both epithelial (transit amplifying (TA) intestinal epithelial cells (TA1 and TA2)) and immune (macrophages, CD8+ T cells and CD4+ T cells). The program was highly expressed in biopsies from UC patients, both the inflamed UC biopsies, where the disease was clinically visible, but also the non-inflamed tissues of the UC patients — suggesting that the MCP also captured molecular markers of the latent disease state. This MCP included multiple UC risk genes previously identified by GWAS, and predicted both UC status (between UC and healthy individuals) and treatment response in an independent “unseen” cohort. Interestingly, the GWAS genes were expressed in different cell types, but their expression was coordinated, a pattern that could only be found as an MCP.
Alzheimer’s disease (AD)
DIALOGUE identified two MCPs spanning multiple cell types that were substantially higher in postmortem brain from AD patients, potentially marking two AD molecular subtypes. These MCPs also demonstrate a strong non-linear increase with age in both frontal cortex (n = 455) and cerebellum (n = 456) of neurologically normal subjects. Like in UC, the MCPs identified by DIALOGUE capture not only the overt but also latent properties of the disease, including disease predisposition and potentially de novo disease subtypes.
Melanoma
DIALOGUE identified an MCP spanning multiple immune cells in melanoma tumors from patients who were resistant to checkpoint inhibitor immunotherapy: the induced part is higher in the responders vs. non-responders, and its repressed part has the opposite pattern. In CD8 T cells the program links CXCR6 overexpression with TCF1 under-expression, consistent with our recent findings that CXCR6 is a pan-cancer marker of T cell dysfunction, which mediates interactions with myeloid cells and is directly repressed by TCF16,7. Interestingly, the program links T cell dysfunction with M2 polarization and APOE repression in macrophage. Indeed, it has been previously shown that ApoE can promote anti-tumor immune responses8,9, with currently ongoing clinical trials to activate ApoE in melanoma patients.
What’s next?
With the rapid increase in single cell and spatial datasets, DIALOGUE will allow comprehensive identification of MCPs across different biological contexts, guiding more rapid mechanistic molecular investigations of multicellular regulation. While we focused here on RNA expression data, DIALOGUE can be applied to any data type to further delineate multi-modal connections and map the vocabulary of MCPs underlying tissue function in health and disease.
One of the future directions we are most excited about is integration of DIALOGUE with Perturb-Seq data10–12, where genetic (e.g., CRISPR or ORF-based) perturbations can be applied in a multiplexed manner with single cell or spatial data as readouts to uncover mechanisms of multicellular regulation and identify new ways to engineer multicellular circuits. A similar application can be done in the context of natural human genetic variation. These combinations could provide powerful approaches to uncover the impact of perturbations on the cell and its neighbors and set the stage for causal inference of gene function in the tissue context, linking genetic causes to single cells to tissue states and phenotypes.
References
- Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018).
- Rodriques, S. G. et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463 (2019).
- Zhu, Q., Shah, S., Dries, R., Cai, L. & Yuan, G.-C. Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data. Nat Biotechnol 10.1038/nbt.4260 (2018) doi:10.1038/nbt.4260.
- Tasic, B. et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018).
- He, S. et al. High-Plex Multiomic Analysis in FFPE Tissue at Single-Cellular and Subcellular Resolution by Spatial Molecular Imaging. bioRxiv 2021.11.03.467020 (2021) doi:10.1101/2021.11.03.467020.
- Jerby-Arnon, L. et al. Pan-cancer mapping of single T cell profiles reveals a TCF1:CXCR6-CXCL16 regulatory axis essential for effective anti-tumor immunity. bioRxiv 2021.10.31.466532 (2021) doi:10.1101/2021.10.31.466532.
- Ruffin, N. & Guerreiro-Cacais, A. O. A pan-cancer signature for dysfunctional T cells. Nature Reviews Immunology 22, 74–74 (2022).
- Tavazoie, M. F. et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 172, 825-840.e18 (2018).
- Ostendorf, B. N. et al. Common germline variants of the human APOE gene modulate melanoma progression and survival. Nature Medicine 26, 1048–1053 (2020).
- Dixit, A. et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853-1866.e17 (2016).
- Frangieh, C. J. et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nature Genetics 53, 332–341 (2021).
- Dhainaut, M. et al. Spatial CRISPR genomics identifies regulators of the tumor microenvironment. Cell (2022) doi:10.1016/j.cell.2022.02.015.
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in