Epigenetic regulation in malignant cancer stem cells
Published in Bioengineering & Biotechnology

Having worked on interdisciplinary research fields with robust cross-disciplinary tangents (for a while now), the epigenetics underlying the origin of cancer has renewed my interest in the field of cancer biology and oncogenetics. Traditionally, the origin and progression of cancer is known as a genetic disease, although with time and modern medicine, biologists discovered the involvement of epigenetic abnormalities alongside genetic alterations during disease onset (Sharma 2010).
In 1942, developmental biologist and geneticist, C.H. Waddington initially defined epigenetics as ‘causal interactions between genes and their products, which bring the phenotype into being’ (Waddington 1942). With time, the definition of epigenetics has evolved to describe a study of heritable changes in gene expression that are independent of changes in the primary DNA sequence, while resulting in changes in the phenotype (Dawson 2017).
In even simpler terms, epigenetic changes are like biological switches that introduce reversible modifications to DNA to regulate genes that can be turned on or off, while attaching to DNA, yet without changing the sequence of the DNA building blocks. Since epigenetic changes underly which genes are turned on/off, they can influence the production of specific proteins in cells. Moreover, the disruption of natural epigenetic mechanisms that broadly occur due to mutations acquired via intrinsic factors or exposure to extrinsic pollutants, dietary changes or environmental influences can trigger altered gene function, to cause malignant cellular transformations (Sharma 2010). The onset of cancer in this way, through mutated epigenetic regulators, can trigger oncogenic cellular reprogramming for uncontrolled self-renewal and the emergence of malignant cancer stem cells (Wainwright 2017). The emergence of cancer stem cells is a direct outcome of the elaborate re-organization of the cancer epigenome (Figure 1).
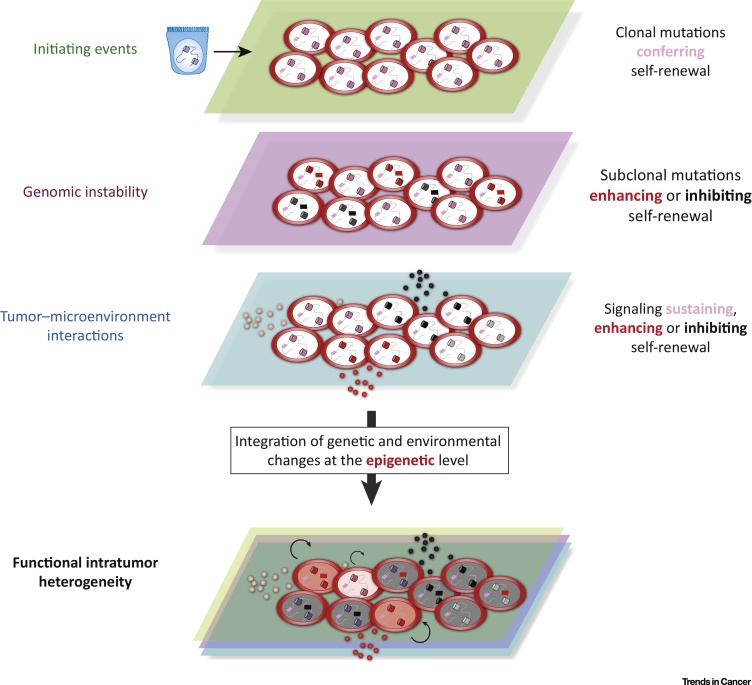
The epigenome – a key to understanding cell fate, gene activity and disease etiology.
During normal mammalian development, epigenetic modifications such as DNA methylation and covalent histone modification (to name a few) are essential for normal growth and gene expression (Table 1). These modifications are collectively termed the epigenome and are key regulators of cell fate and gene activity (Mikkelsen 2007). While healthy embryonic stem cells can maintain a plastic epigenome with room for change during developmental processes, differentiated tissue can display a relatively restricted structure that is stably maintained via cell division. The distinct patterns underlying modifications across diverse cell types, including cancer cell lines serve as a guardian of cellular identity (Jones 2007). The fact that these epigenetic aberrations are potentially reversible unlike genetic mutations – with potential to restore their normal state via epigenetic therapy further highlight the promising and therapeutically relevant impact of these findings.
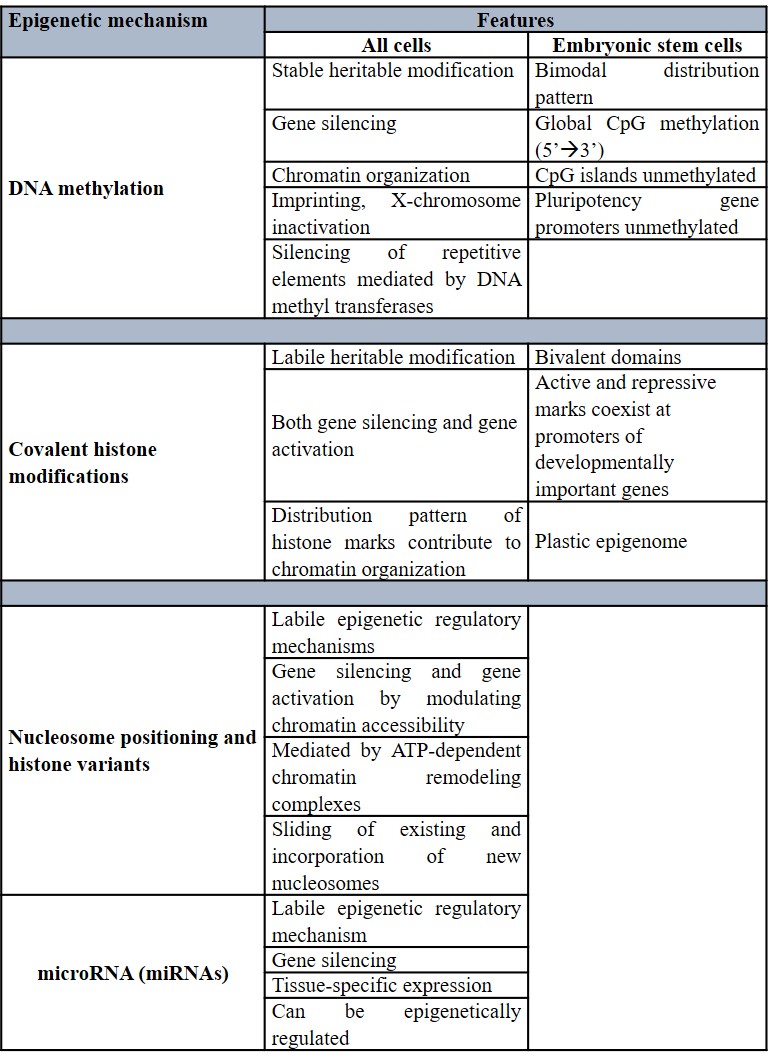
Epimutations of cancer initiation and progression – oncogenic cellular reprogramming
In 1971 geneticist Alfred Knudson proposed a two-hit hypothesis for cancer initiation. Where tumor suppressor genes that act as brakes to stop cells from transforming to malignant cancer cells, while also attenuating malignant cells from undergoing uncontrolled self-renewal, may themselves undergo mutation (Knudson 1971). However, such mutations are recessive in nature, and a cell can therefore only truly become cancerous by acquiring a two-hit mutation in both tumor suppressor genes of a single cell.
The epigenetic alterations promoting cancer initiation and tumorigenesis are many and varied (Figure 2). Example events include hypomethylation that increase genomic instability to activate proto-oncogenes (Feinberg 1983), and site-specific hypermethylation that contributes to tumorigenesis by silencing tumor suppressor/DNA repair genes such as p16, MLH1, and BRCA1 (Baylin 2005). Post-translational modifications such as lysine methylation have revealed new complexities in facilitating signal transduction pathways with critical regulatory roles in cancer promoting pathways (Bao 2018), bringing us to a very interesting biomechanism of hypoxic regulation of cancer stem cells, and its pathological relevance.
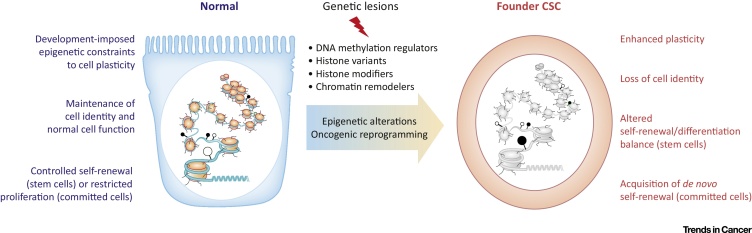
A closer look: Epigenetic crosstalk in a hypoxic microenvironment to regulate cancer stem cell mobility.
In 2019, three scientists received the Nobel prize in physiology or medicine for their discoveries of how cells sense and adapt to oxygen availability (Chopra 2020). Oxygen-sensitive enzymes and cellular machinery can coordinate to turn-off major regulatory protagonists at play such as ‘hypoxia-inducible factor’ proteins – pivotal to cancer pathways. Hypoxic conditions primarily underly the etiology of several cancer types and solid tumorigenesis, including colorectal cancer and glioblastoma, an aggressive brain cancer (Rawluszko 2013, Bao 2018).
Hypoxia inducible factor (HIF-1α) is a major player and master transcriptional regulator during the adaptation of cancerous tissue to hypoxia, and is known to drive the mobility of cancer cells. This protein is negatively regulated by a family of prolyl hydroxyl enzymes including PHD1, PHD2, and PHD3 (encoded via the egg-laying defective nine (EGLN1-to-3) genes), as well via factor inhibiting hypoxia inducible factor (FIH) (Bao 2018).
During normoxia or normal oxygen levels, the oxygen sensor PHD enzymes catalyze hydroxylation of the oxygen-dependent degradation domain located on HIF-1α. Rendering the protein identifiable to a ligase complex containing von Hippel-Landau tumor suppressor protein, for targeted degradation of the molecule, to prevent HIF-1α from interacting with its co-activators. During hypoxia or low oxygen levels (typical to tumor microenvironments), the family of oxygen sensor enzymes (PHDs) are downregulated for HIF-α driven cancer progression in primary cancerous tissue. This epigenetic mechanism is well represented during HIF-1α driven migration of glioblastoma cancer cells (Bao 2018).
Deeper introspection: peering into the epigenetic cellular machinery.
The HIF-1α dependent migration of glioblastoma stem cells can also be regulated by lysine methyltransferase enzymes G9a and G9a-like protein (GLP) that directly bind to the alpha subunit of HIF-1α to impart methylation of the protein. This methylation can suppress HIF-1α transcriptional activity in glioblastoma cells inhibiting the function of the hypoxic protein by reducing its transactivation domain function (Shinkai 2011). This pathway is not an outcome of increased HIF-1α protein degradation as detailed earlier, instead it presents a new avenue for low-oxygen signaling, highlighting the role of G9a. During hypoxia in glioblastoma, G9a downregulation can also be inversely correlated with an angiogenesis-regulating protein PTGS1 expression, resulting in the survival of patients, although chronic hypoxic conditions can cause continued tumorigenesis (Bao 2018).
In this way, lysine methylation drives cancer-promoting pathways and presents druggable targets for therapeutic design (McGrath 2015), therefore the role of G9a in hypoxia is assumed to be larger than what is currently known (Chopra 2020). Incidentally, the oxygen sensor PHDs can manipulate all these factors, including the hypoxia inducible G9a and GLP proteins, by suppressing lysine methyltransferase activity (Figure 3). Reduced PHD expression on the other hand correlates with DNA methylation in its promoter regulatory region. Which brings us to an important question; how do we dissect some of these enzyme-substrate networks in the lab?
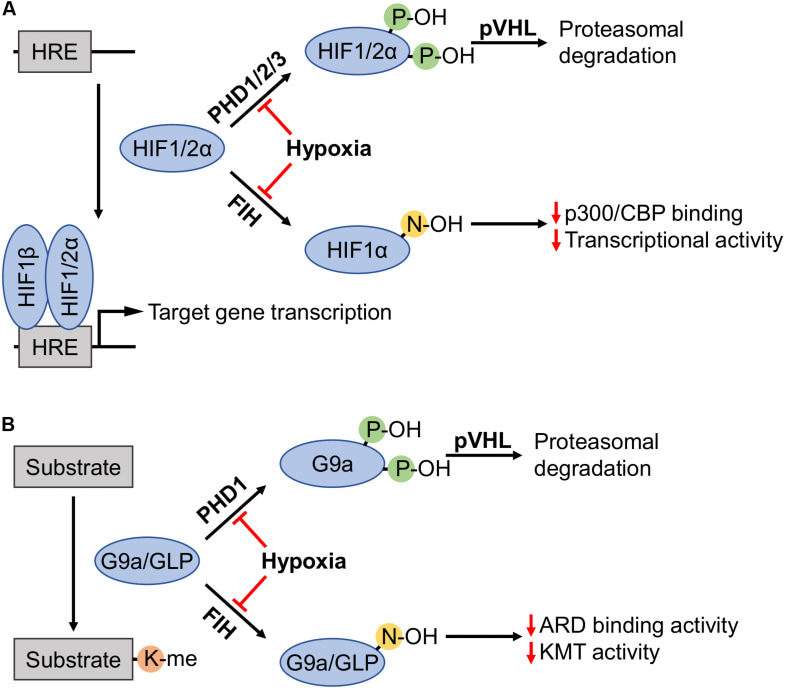
Regulation of HIF1α subunits and G9a/GLP via prolyl and asparaginyl hydroxylation. (A) In the presence of oxygen, PHDS catalyze the proteolytic degradation of HIF-1α. (B) Much like HIF-1α regulation, PHD1 can induce the catalytic degradation of G9a/GLP (Chopra 2020).
Highly regulated localized epigenetic experiments
Very briefly, it is possible to bioengineer a malignant stem cell line with an EGLN gene knockout to yield cells with inactive PHD protein functionality, for comparative assessments of its biology relative to native malignant stem cells vs. native stem cells, under conditions of induced hypoxia (Figure 4). These experiments would ideally follow preliminary studies conducted with malignant stem cell lines vs. normal stem cell lines to examine the expression of PHD enzymes under hypoxia and normoxia. The functional assays can parallelly investigate the expression of G9a/GLP lysine methyl transferases under varied culture conditions in vitro.
These experiments can be conducted using microfluidic organ-chips to recreate a highly regulated hypoxic tumor-like microenvironment in vitro and observe the mobility or inhibition of cancer cells and their dynamics of self-renewal in a localized environment. Organ-chips need no further introduction on this post, as I reference previous notes of the concept from December 2016 and December 2022; the latter similarly on bottom-up engineering a cell signaling pathophysiological pathway on a chip.
The microfluidic instrument can provide a microphysiological window to regulate in vitro biological conditions throughout the course of an experiment, to ask and answer fundamental questions of hypoxia-related epimutations in a localized microenvironment. These highly regulated methods discussed herein are localization experiments, in contrast it will be necessary to consider multiple variables during systemic experiments in animal models. Preliminary outcomes of the experimental design can lead to a series of successive functional assays to investigate hypoxia-related pathways of the epigenome during cancer initiation and cell mobility/self-renewal among varied cancer stem cell lines.
In this way, genetic alterations and altered patterns of epigenetic expression are key features of cancer initiation (neoplasia) and progression with stem cell contributions. This brief essay outlined some of the fundamental discoveries in the field with an overarching aim to investigate strategies of epimutations for epigenetic therapy to target cancer stem cells (Jones 2019), and control cancer.
Header Image: How Epigenetic Marks can Change the Genome - The Scientist Infographic: How Epigenetic Marks Can Change the Genome | TS Digest | The Scientist (the-scientist.com)
References
- Sharma S. et al. Epigenetics in cancer, Carcinogenesis, doi: 10.1093/carcin/bgp220
- Waddington C.H., Canalization of development and the inheritance of acquired characteristics, Nature, doi: https://doi.org/10.1038/150563a0
- Dawson M. A., The cancer epigenome: Concepts, challenges, and therapeutic opportunities, Science, doi: 10.1126/science.aam7304
- Wainwright E. et al. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity, Trends in Cancer, doi: doi: 10.1016/j.trecan.2017.04.004
- Mikkelsen T. S. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells, Nature, doi: 10.1038/nature06008
- Jones P.A., et al. The epigenomics of cancer, Cell, doi: 10.1016/j.cell.2007.01.029
- Knudson A.G. et al. Mutation and cancer: statistical study of retinoblastoma, PNAS, doi: https://pubmed.ncbi.nlm.nih.gov/5279523/
- Feinberg A.P. et al. Hypomethylation distinguishes genes of some human cancers from their normal counterparts, Nature, doi: 10.1038/301089a0
- Baylin S. et al. DNA methylation and gene silencing in cancer, Nature Clinical Practice Oncology, doi: 10.1038/ncponc0354
- Bao L. et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration, Nucleic Acids Research, doi: 10.1093/nar/gky449
- Chopra A. et al. Hypoxia-Inducible Lysine Methyltransferases: G9a and GLP Hypoxic Regulation, Non-histone Substrate Modification, and Pathological Relevance, doi: 10.3389/fgene.2020.579636
- Rawluszko A. et al. Expression and DNA methylation levels of prolyl hydroxylases PHD1, PHD2, PHD3 and asparaginyl hydroxylase FIH in colorectal cancer, BMC Cancer, doi: https://doi.org/10.1186/1471-2407-13-526
- Shinkai Y. et al. H3K9 methyltransferase G9a and the related molecule GLP, Genes & Development, doi: 10.1101/gad.2027411
- McGrath J. et al. Targeting histone lysine methylation in cancer, Elsevier, doi: 10.1016/j.pharmthera.2015.01.002
- Jones P. A. et al. Epigenetic therapy in immune-oncology, Nature Reviews Cancer, doi: https://doi.org/10.1038/s41568-019-0109-9
I am an interdisciplinary researcher with a strong commitment to bioengineering, biochemistry, organ-chips and molecular biology. As well as biomechanics and biomineralization in the broader context of medicine. I completed my PhD at the University of Sydney Australia in December 2016, and travel often, find me on Twitter and irl.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in