The paper in Nature Communications is here: https://go.nature.com/2Qcycry
The antiviral activity of type I interferons is indisputable, with few exceptions. Throw some interferon on cultured cells, and they become impenetrable to viral invasion. The ability of interferons to confer a protected, antiviral state is dependent on transcriptional activation of hundreds of genes, many of which are unique to this virus-targeted program.
In the Schoggins lab, we are interested in dissecting the interferon response, one protein at a time. Screens of individual interferon-stimulated genes (ISGs) against a panel of diverse viruses revealed the modularity of the interferon response—certain ISGs inhibit distinct groups of viruses. In the same screens, we also identified a subset of ISGs that promoted the infectivity of one or more viruses.
The concept of single ISGs enhancing viral infection while belonging to a canonically antiviral program is intriguing and has some precedence in the literature. Suppressor of cytokine signaling (SOCS) family proteins, which are interferon-inducible, restrict the interferon response by targeting STAT1-mediated signaling, but our screens were largely performed in STAT1-deficient cells. There are also examples of antiviral proteins that have been ‘hijacked’ by a virus- in essence, repurposed to promote a step in the viral cycle. And then there is lymphocyte antigen 6 complex, locus E (LY6E), which enhanced infectivity by unrelated viruses in our screens, including multiple flaviviruses, a couple of alphaviruses, human immunodeficiency virus, and influenza A virus (IAV). Could all these viruses be hijacking the same protein? As explained below, we’re not convinced that a simple hijacking mechanism tells the whole story of what LY6E is doing.
The earlier literature on LY6E didn’t point to a mechanism behind our phenotype, which was rather frustrating at the time. Several studies in the mid-90s demonstrated a role for murine Ly6e in modulating T cell receptor (TCR) signaling- but our fibroblasts didn’t express TCR! A couple of more recent studies established an essential role for murine Ly6e in proper placental formation, likely due to its interaction with syncytin A which is required for fusion of syncytiotrophoblast layer I. However, our cell lines are not placental in origin, and most of our enhanced viruses do not classically infect placenta. While multiple studies implicated the LY6 locus as important for viral susceptibility, we didn’t have much to start with for a mechanism other than a strong structural resemblance to snake neurotoxins.
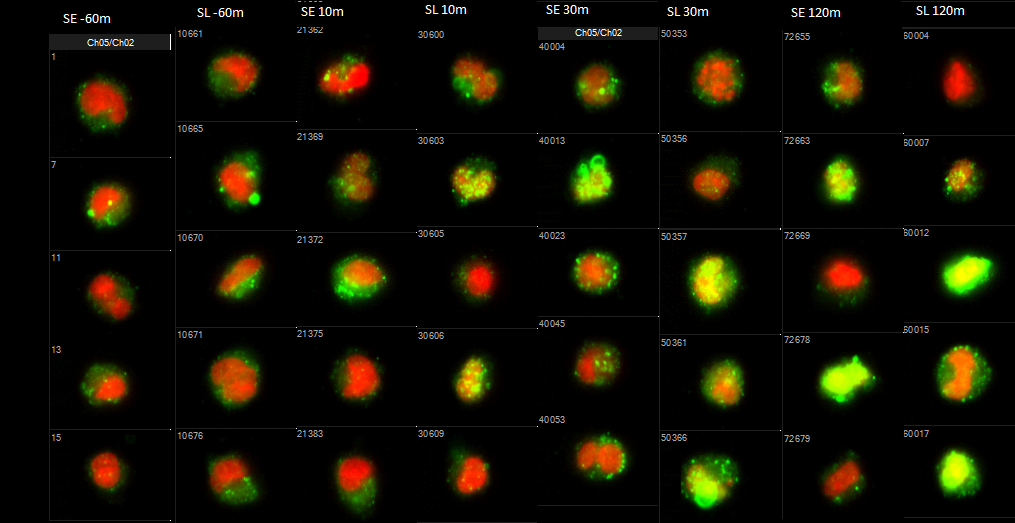
Lucky for us, we established that LY6E promotes viral entry within the first year of the study. Figuring out how entry was affected took the next four years. Like most graduate students, I ran head-first into more dead-end hypotheses than I would normally like to admit. Collaborating became key to getting out of the LY6E maze, with Nick Rinkenberger deserving a large part of the credit here. While I was more interested in exploring potential host proteins and signaling pathways modulated by LY6E, Nick spent the past few years establishing protocols for studying IAV internalization and endosomal escape for his study of another ISG, MCOLN2. He tested whether LY6E enhanced either of these critical steps in IAV entry and lo and behold, it had absolutely no effect on internalization or endosomal escape.
However, these two pieces of negative data made me recall a series of acid bypass experiments I performed back in 2015 on the advice of Dr. Sara Cherry when she visited UTSW as a seminar speaker. The concept behind the acid bypass experiment is that if your protein promotes endosomal entry of virus, treating virus-bound cells with an acidic media to induce fusion at the plasma membrane would lead to a loss the viral enhancement phenotype. I did this experiment many times, and always saw LY6E-mediated enhancement even with acid treatment. Thinking that I was doing something wrong, I filed the acid bypass results with my growing pile of presumed failed experiments and moved on. However, in the context of all our other IAV experiments showing that LY6E affects a step after endosomal escape but before nuclear translocation, we realized that it must be promoting uncoating. It all fell together: acidic treatment leads to fusion of the viral envelope with the plasma membrane, leaving the viral capsid protecting the genome. Loss of the capsid proteins via uncoating is the last step before viral ribonucleoproteins translocate into the nucleus to replicate. We were then able to obtain the anti-M1 antibody HB64 from Dr. Andrew Pekosz and perform the previously published IAV uncoating experiment that completed our current study of how LY6E enhances viral infection.
The biggest question remaining for LY6E is physiological relevance. We show in our study that LY6E is decently conserved amongst 7 mammalian families. Our viral enhancement phenotype is also maintained by orthologs sharing as little as 60% identity, indicating conservation across evolution and potentially an important role in antiviral immunity. Understanding the contribution of LY6E to the overall antiviral response requires studying it in the context of an intact immune system by using an animal model; unfortunately, ablation of whole body murine Ly6e is embryonic lethal. We are hopeful that conditional Ly6e knockout models will help us study Ly6e in a more physiological context and ascribe a more specific role to this interferon-inducible protein than ‘virus companion.’
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in