Fitting mutational signatures: benchmarking and more
Published in Cancer, Protocols & Methods, and Mathematics
Mutations do not appear in the genome at random. The patterns of their appearance, referred to as "mutational signatures", have been studied for a long time. In the 1990s, various types of mutations of the tumor suppressor gene TP53 have been associated with prior exposure to environmental carcinogens1. Thanks to next-generation sequencing and the introduction of a robust mathematical framework for mutational signatures by the group of Michael Stratton at the Wellcome Sanger Institute2, mutational signatures have gradually become a widely used tool in genomics. They can help determine the absolute timing of mutations3, uncover biological processes that take place in living tissues4, and serve as prognostic or therapeutic biomarkers5. The reference catalog of mutational signatures has gradually grown from 22 signatures in 2013 to more than 160 signatures in 2024 (see the standard COSMIC catalog of mutational signatures).
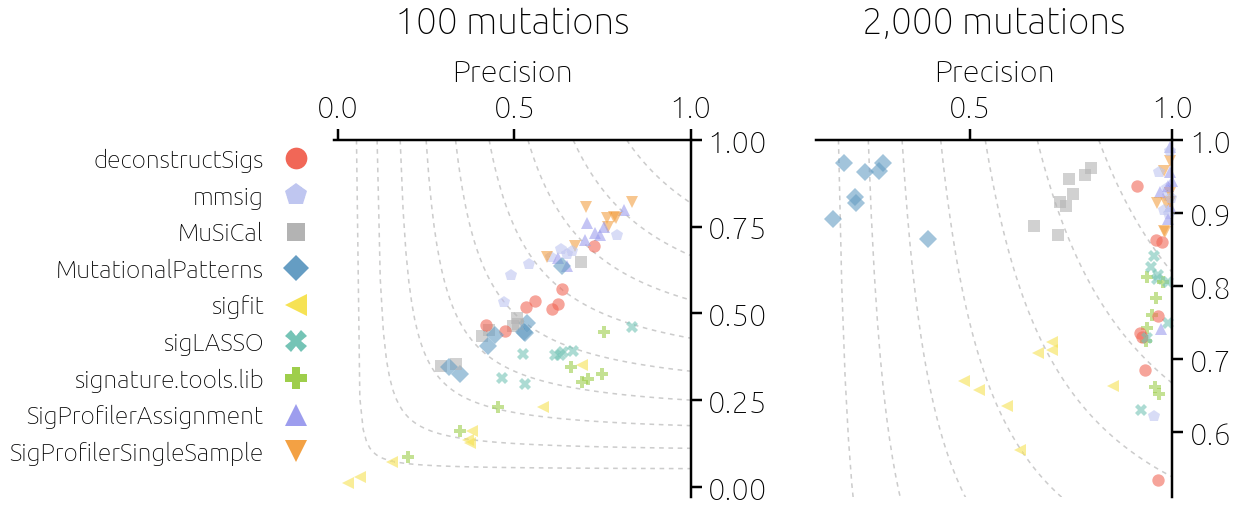
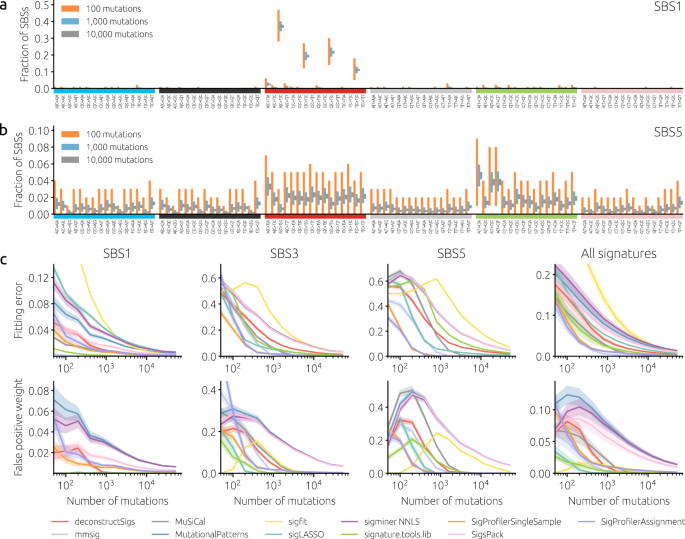
Mutational signatures have become a standard tool, so when we worked on a genomic project in 2021, it was clear that we wanted to analyze their activity in our samples. It was an interesting project centered around a large cohort of patients with squamous cell carcinoma of the head and neck with lymphatic metastasis. The patient tissues were analyzed by whole exome sequencing, which generally yields far fewer mutations than whole genome sequencing. In our case, the median number of mutations in a sample was around 100, which is the range at which signature analysis becomes difficult. While 100 mutations may seem like a lot, for detecting mutational signatures it is not. The problem is that the most common scheme to define mutation patterns is based on single based substitutions. While there are only six possible substitutions (if we neglect the strain), they are considered together with their immediate adjacent bases. As each adjacent base has four different possibilities, this gives a classification of mutations in 6 * 4 * 4 = 96 different classes. When 100 mutations from a sample are distributed among approximately the same number of classes, the number of mutations of each type is small. This directly leads to large noise in the data and, in turn, the difficulty in accurately estimating signature activity in the sample (Figure 1).
Our genomic project was important and we wanted to do the signature analysis in the best way possible. However, many tools were available for analysis and there was no clear consensus on which performs best. There was a recent preprint comparing tools for discovering new signatures (now published6) but we wanted to mainly fit known signatures to our samples (we also checked our samples for new signatures and found none). This led us to the idea of an independent study where we would extensively benchmark the available tools for fitting mutational signatures. Finding time for a new project is always difficult but eventually, we did set up a model for realistic synthetic data on which the tools were to be tested and collected more than ten common tools for evaluation. We designed various ways how tool performance can be evaluated - using various error metrics as well as using the produced activity estimates in downstream analysis, and some months later, the paper was ready for submission (Figure 2).
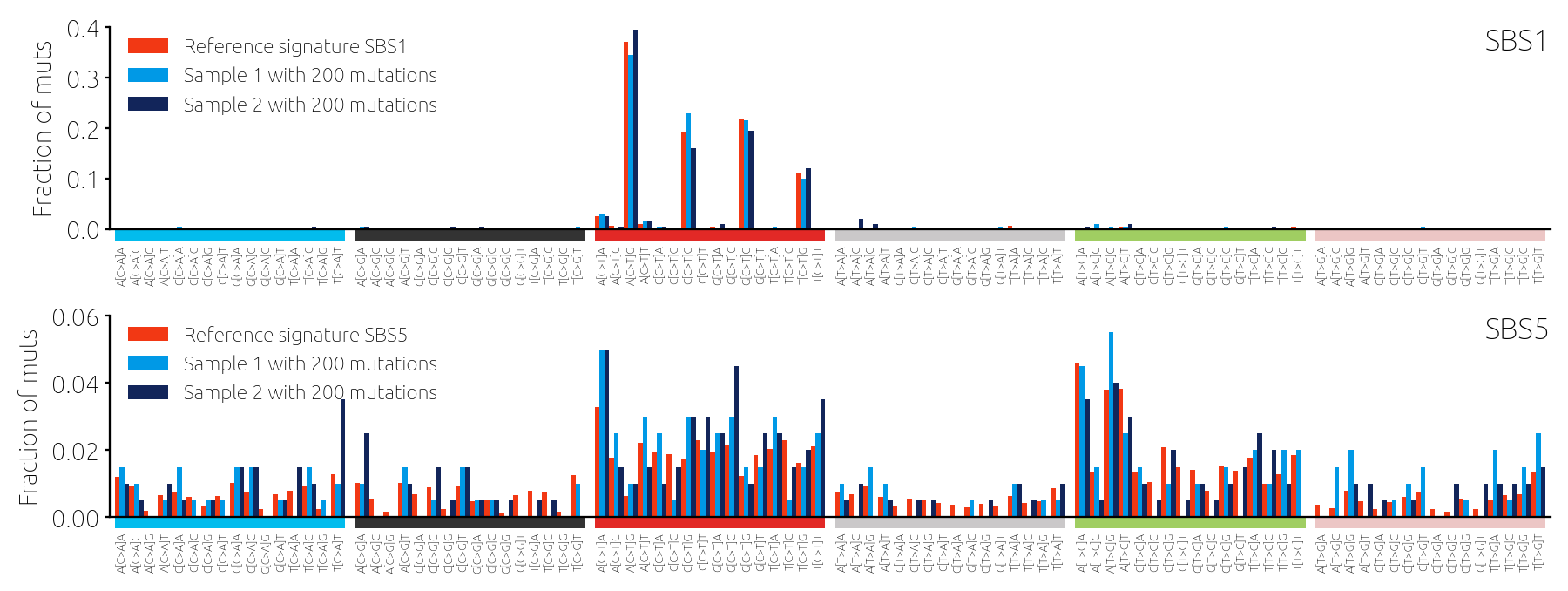
The reviewer reports were very positive which was a nice surprise to us because in academia, finding ways to criticize others is often too easy. The support of the editor and the reviewers also confirmed our initial decision to invest time in this "side" project. However, not everything was great as the reviewers shared the opinion that we should use some real data to support our results, which have been based exclusively on synthetic data so far. This seemed like a superfluous requirement because we felt that we invested a lot of effort in our model for synthetic data and real data will only introduce difficulties without bringing anything beneficial. At the same time, it was a natural suggestion, and so we decided to give it a try. The results did not please us - they were in an important way different from everything that we saw for the synthetic data. For the synthetic data, when mutations were plentiful, all tools (almost) produced highly accurate results that differed very little from one tool to another. For the real data, however, large differences between tools were also observed in samples with many mutations.
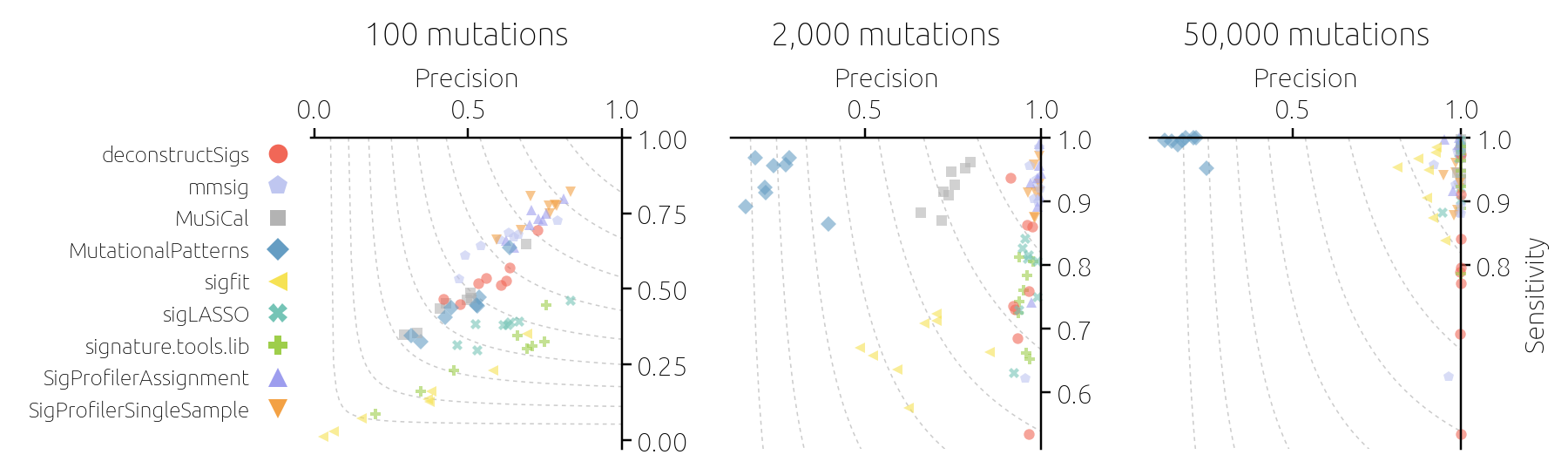
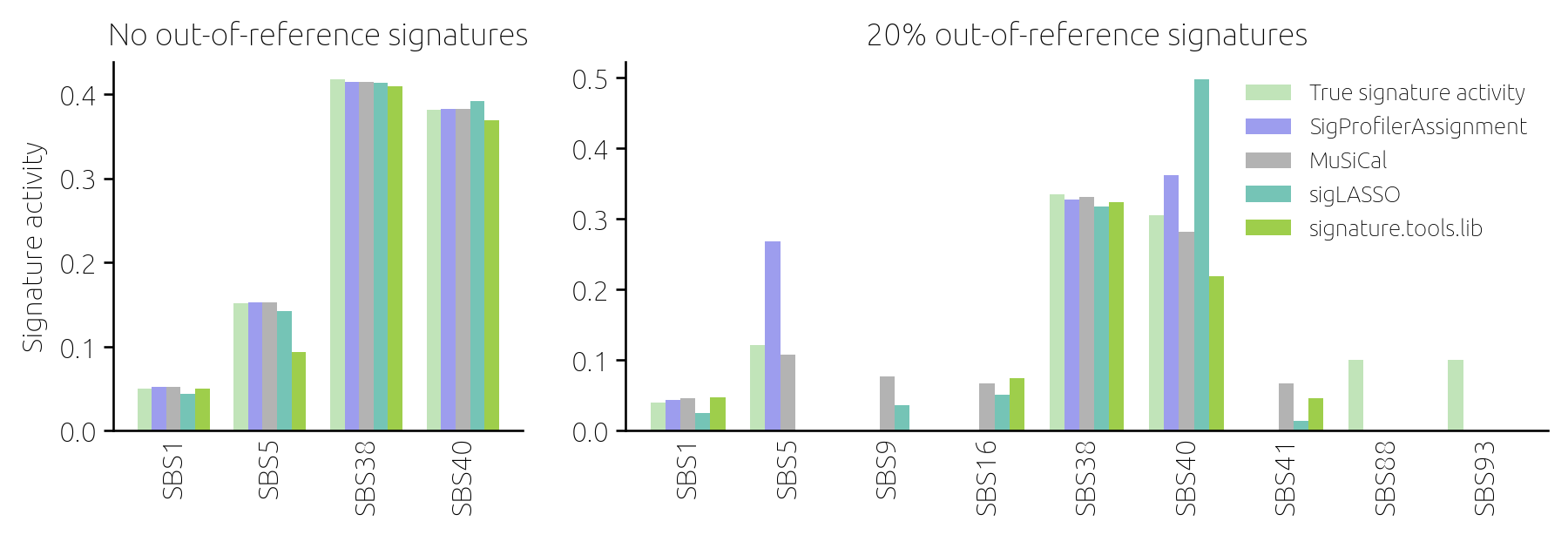
At that point, it was the versatility of the model for synthetic data that came to the rescue. We varied the model assumptions until we finally came across one that allowed us to obtain results similar to those found using real data. We found that when a small fraction of mutations in a sample are due to signatures that are not in the reference catalog, the tools struggle and their estimates disagree (Figure 3). This became one of the key findings of the paper. We not only found which tools to fit mutational signatures perform best but also identified the activity of unknown signatures as the biggest challenge to accurate signature analysis. Following the reviewers' nudge really paid out this time.
References
- Hollstein, M., Sidransky, D., Vogelstein, B., & Harris, C. C. (1991). p53 mutations in human cancers. Science, 253(5015), 49-53.
- Nik-Zainal, S., Alexandrov, L. B., Wedge, D. C., Van Loo, P., Greenman, C. D., Raine, K., ... & Stratton, M. R. (2012). Mutational processes molding the genomes of 21 breast cancers. Cell, 149(5), 979-993.
- Leshchiner, I., Mroz, E. A., Cha, J., Rosebrock, D., Spiro, O., Bonilla-Velez, J., ... & Rocco, J. W. (2023). Inferring early genetic progression in cancers with unobtainable premalignant disease. Nature Cancer, 4(4), 550-563.
- Koh, G., Degasperi, A., Zou, X., Momen, S., & Nik-Zainal, S. (2021). Mutational signatures: emerging concepts, caveats and clinical applications. Nature Reviews Cancer, 21(10), 619-637.
- Levatić, J., Salvadores, M., Fuster-Tormo, F., & Supek, F. (2022). Mutational signatures are markers of drug sensitivity of cancer cells. Nature Communications, 13(1), 2926.
- Islam, S. A., Díaz-Gay, M., Wu, Y., Barnes, M., Vangara, R., Bergstrom, E. N., ... & Alexandrov, L. B. (2022). Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genomics, 2(11), 100179.
I work on problems in biostatistics, genomics, complex systems, complex networks, and information filtering.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in