Major histocompatibility complex (MHC) class I molecules are cell surface proteins that act a bit like a cellular alarm system alerting the immune system to the existence of foreign substances and triggering an initial immune response. If, for example, MHC molecules float by a viral peptide that has a high affinity it will bind to the peptide and then present it to cytotoxic T cells on the cell’s surface. The T cells are able to detect foreign peptide/MHC combinations and induce apoptosis in the cell.
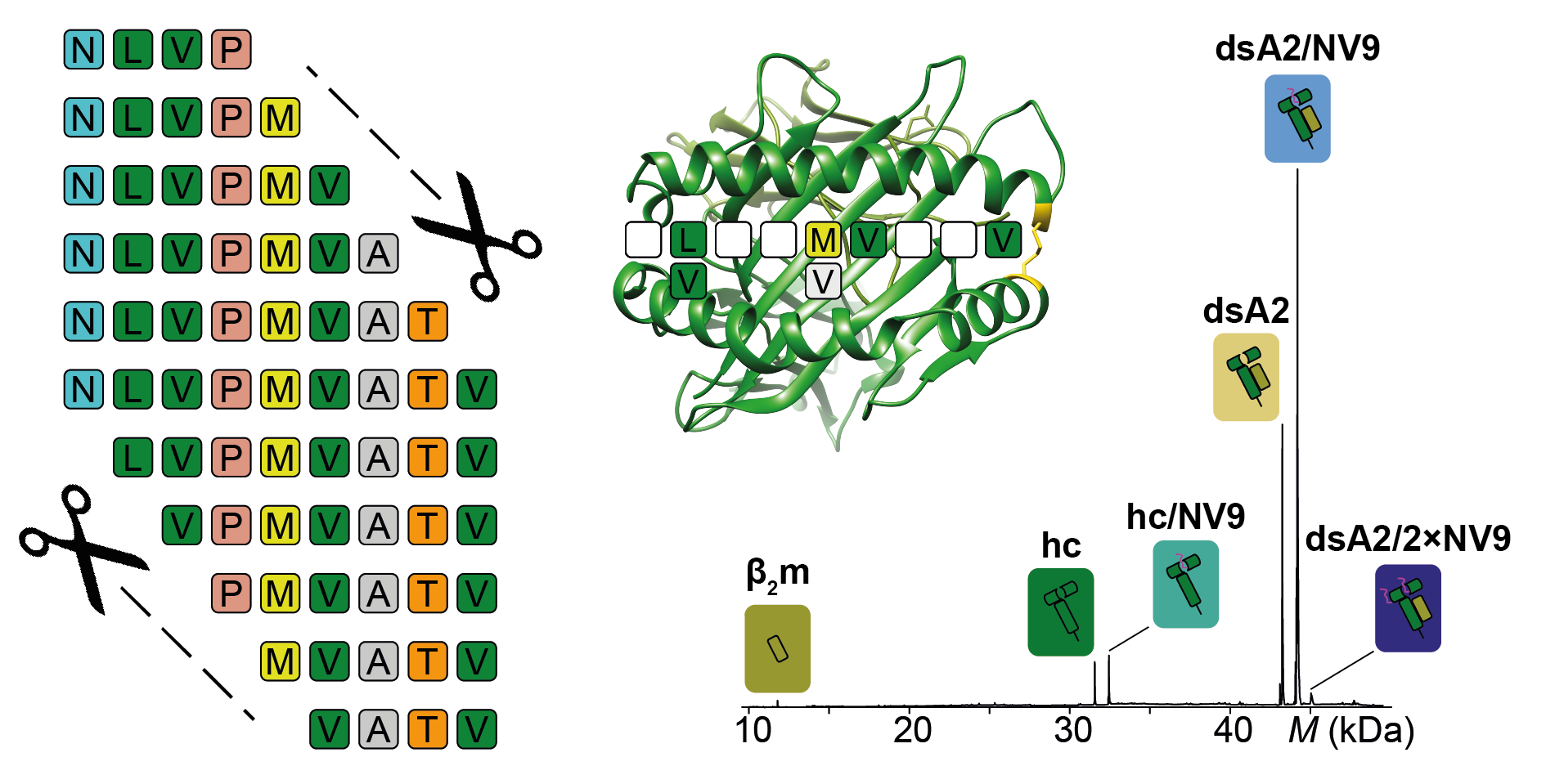
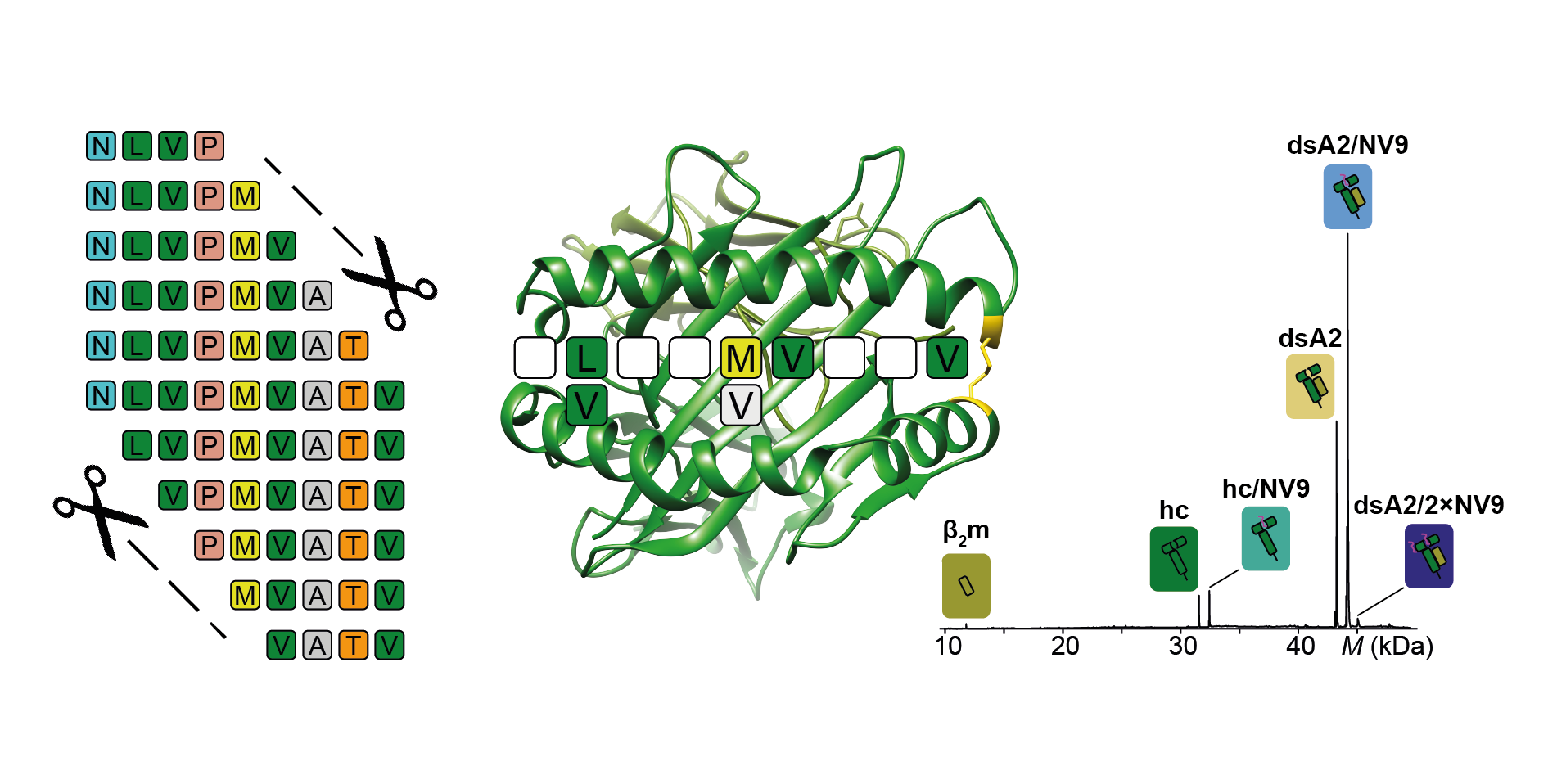
In general, we would like to understand which peptides bind to MHC molecules so that we can predict the immune systems’ response to individual peptides. As MHC molecules are complexes formed by two subunits, which fall apart when no peptide is attached, it is difficult to look at the structural features of the complex. In a previous paper, we demonstrated a method for stabilizing an MHC molecule with a system bridge at the outer peptide binding pocket. These empty disulfide-stabilized class I molecules (dsMHC) can be examined bound to peptides using native mass spectrometry (nMS), which detects all the different mass species present in a solution.
With MHC stabilized, we were excited to start analyzing MHC-peptide binding with many different peptides to determine what features are important for sufficient binding. After determining the nMS “sweet spot”, the voltage needed to generate optimal resolution for the sample, we began looking at different peptide lengths and sequences as well as peptides with modified termini.
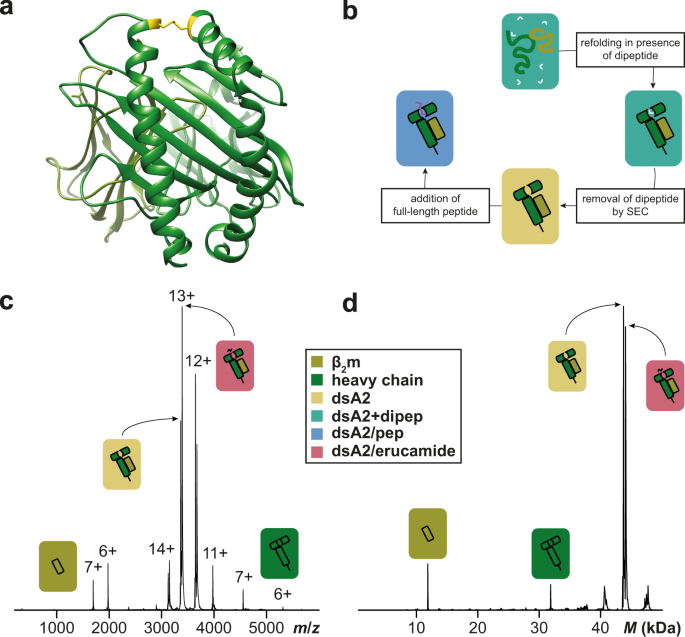
Our results were, however, not what we had expected. We kept finding tiny traces of an unknown molecule other than the peptide that was not only consistently binding with the MHC molecules, but also occupying 100% of the binding pockets when no peptide was present. We were baffled and could not figure out what this unknown molecule could be.
Over the next couple of years, we continued to ask colleagues at conferences and our collaborators for help in identifying our mystery molecule but everyone was stumped. Luckily, we reached out to Thomas Dülcks from University of Bremen who specializes in MS of smaller molecules. He was able to identify our mystery molecule as erucamide, a softener used in plastics, that was most likely coming from our test tubes.
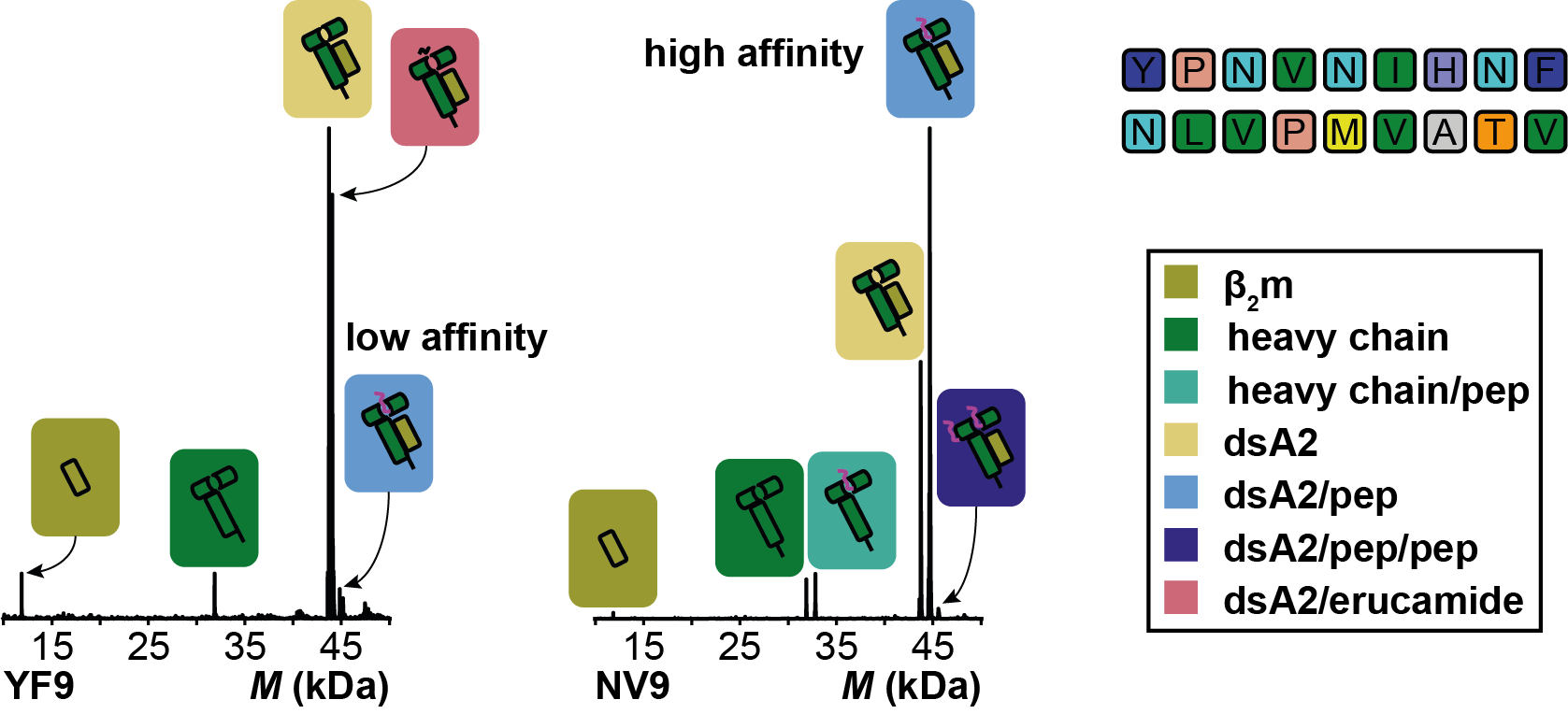
Once our mystery molecule was identified, we realized that we could turn erucamide’s high affinity binding into something beneficial. Given that erucamide occupies 100% of the binding pocket, we could add peptides to erucamide bound MHCs and determine the peptides affinity based on the fraction of erucamide’s replacement. Our mystery molecule had suddenly given us the basis for developing a novel high-throughput peptide screen for MHC class I epitopes. We of course then went on to test this new method and with the help of CSSB’s Protein Characterization facility we could also validate the method’s accuracy.

Ultimately, the identification of high affinity virus- or tumor-specific peptide epitopes would contribute to the development of peptide vaccines, which have not yet been approved for human use. Unlike conventional vaccines and RNA vaccines, peptide vaccines would be stable and therefore have a significant advantage as they would not require specific storage conditions. This is extremely important as there is a high need for vaccinations in tropical and hot climates with limited medical infrastructure.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
DNA repair and human disease
Publishing Model: Hybrid
Deadline: Oct 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in