Getting organised: Structural transformations using energy dissipation
Published in Chemistry

Nature uses the constant input and transformation of energy to modulate the form and function of its chemical systems. For example, in natural tissues such as collagen, energy conversion is used to vary the elasticity and robustness of fibres for integration into different tissue types. Similarly, networks of fibronectin consume and transform chemical energy to create a reconfigurable mechanical support for cells and for signalling pathways within the extracellular space. Unlike these remarkable biological materials, man-made fibres do not typically exhibit complex and dynamic behaviours and instead require irreversible synthetic modifications to change their mechanical properties. To create life-like materials with hierarchical structures that can be varied on-demand, new chemical procedures are required.
Due to its predictable assembly and molecular recognition properties, DNA is an attractive construction material for generating new fibrous architectures. DNA is also inherently dynamic due to the non-covalent interactions that govern its self-assembly into higher order structures, making it an ideal candidate to produce biocompatible materials with tuneable properties. The very chemical interactions holding a double helix of DNA together – hydrogen bonding and pi-pi interactions – can be reprogrammed to recognise unnatural nucleotide analogues. The resulting structures, taking inspiration from Watson-Crick base pairing, can produce new libraries of architectures for non-covalent material construction.
Our group recently showed that DNA can be paired with a commercially available small molecule – cyanuric acid – to produce one-dimensional supramolecular polymers. When mixed at room temperature, cyanuric acid (whose structure closely matches that of the natural nucleotide thymine) “base pairs” with poly(adenine) DNA strands to form triple helices that polymerise into micron-length fibres that interconnect and grow to form tangled, interwoven networks. This process is uncontrolled, resulting in structural defects throughout the material and limiting the applicability of these fibres in tissue engineering applications.
We hypothesised that the structural properties of our DNA-small-molecule fibres could be improved by employing bio-inspired strategies: that the consumption and dissipation of energy would alter the assembly pathway of individual fibres, modulating the local and global structure of our assemblies. To achieve this goal, we combined our DNA-small-molecule material with a photochemical system that relies on irradiation with a smartphone camera light. When activated, protons released from this system unravel our DNA fibres, generating a high-energy DNA duplex state. This process breaks up our disordered fibre networks and sequesters the DNA monomers in to an inert, inactive form. When the light is switched off, the system relaxes, monomers are slowly release from their storage depot intermediate, and our fibres reassemble.
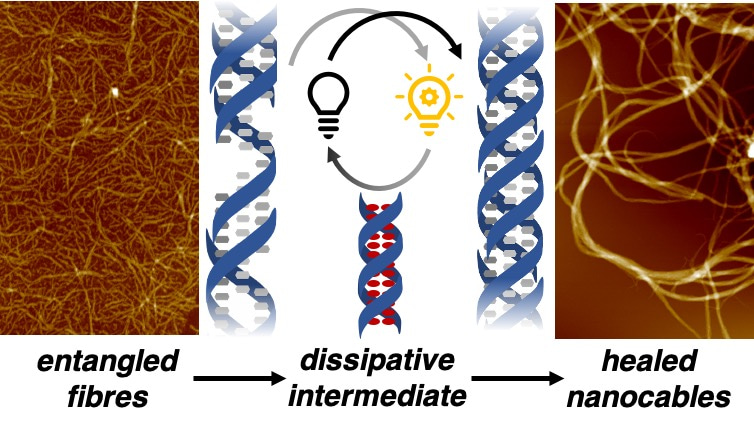
Unexpectedly, we observed that as monomers are released from our dissipative intermediate, the initial intermeshed product is not reformed: instead, individual fibres aggregate along their axes of polymerisation, generating thick “nanocables”. These superstructures were found to have improved mechanical properties and thermal stabilities due to their slower, more controlled growth, demonstrating how an off-pathway process can be used to manipulate the properties of materials, analogous to how Nature controls the function of biological tissues.
Using state-of-the-art fluorescence microscopy techniques, we observed that the slow release of monomer units from the irradiated state switched the mechanism of self-assembly at the single-fibre level. Initially formed by a combination of monomers adding one-by-one and large oligomer fragments adding, the activation of our dissipative cycle selects for a polymerisation mechanism wherein monomers associate one-by-one only. We hypothesise that the selectivity of this mechanism following energy dissipation is due to a kind of ‘auto-titration’ of monomer DNA strands, which modulates the concentration of monomers available for polymerisation according to the kinetic relaxation profile of the photoacid.
Switching the assembly pathway of polymerisation impacted local fibre structure. We developed a simple assay to test for heterogeneity in our DNA fibres using single-molecule fluorescence microscopy. We observed that fibres formed using our dissipative strategy have fewer structural defects than those grown without cycle activation. Our more ‘perfect’ individual fibres are thus prevented from branching, and are instead encouraged to aggregate along their axis of polymerisation, giving rise to robust and organised cables.
While single-molecule fluorescence techniques have been widely used to study biological systems, this study marks the first direct observation of the different mechanisms of supramolecular polymerisation, and the first optical assay developed to probe the heterogeneity of supramolecular polymers. We anticipate that these new methodologies will be broadly applicable to the study of both natural and synthetic supramolecular polymers and may provide important insight into how Nature controls the properties of its functional tissues, enabling scientists to produce more dynamic and tunable materials. Our ability to control the hierarchical organisation of nanomaterials using chemical stimuli under kinetic control marks a milestone in the development of out-of-equilibrium systems for materials organisation.
Follow the Topic
-
Nature Chemistry

A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in