Graph deep learning detects contextual prognostic biomarkers from whole slide images
Published in Bioengineering & Biotechnology

The recent development of deep learning in image analysis advanced computational pathology. Currently, the segmentation and classification of the different cell types in a high-resolution whole slide image (WSI) are highly accurate, and predicting the oncogenic variant, gene expression, or even origin of metastasis is possible. However, deep learning-based pathology usually used the small-patch images obtained from splitting the WSI as the input data. Thus, the deep learning model focuses on local features such as morphological changes or growth patterns of tumor cells. However, pathologists review local features within the context because some pathological features are explained differently according to the surrounding tumor microenvironment (TME). For example, immune cells have totally different roles whether they interact with tumor cells or stromal cells. Therefore, we need a new method that extracts meaningful contextual features reflecting the surrounding environment together with the local features.

To consider such contextual features in gigapixel WSIs, we introduced tumor environment-associated context learning using graph deep learning (TEA-graph), which is a graph neural network (GNN)-based method that analyzes the contextual histopathology features of gigapixel WSIs in a semi-supervised manner. TEA-graph represents the WSI into a memory-efficient, graph-like structure to efficiently reflect the relationships between the local features using the GNN method. In addition, TEA-graph extracts pathological context features through an interpretable GNN model.
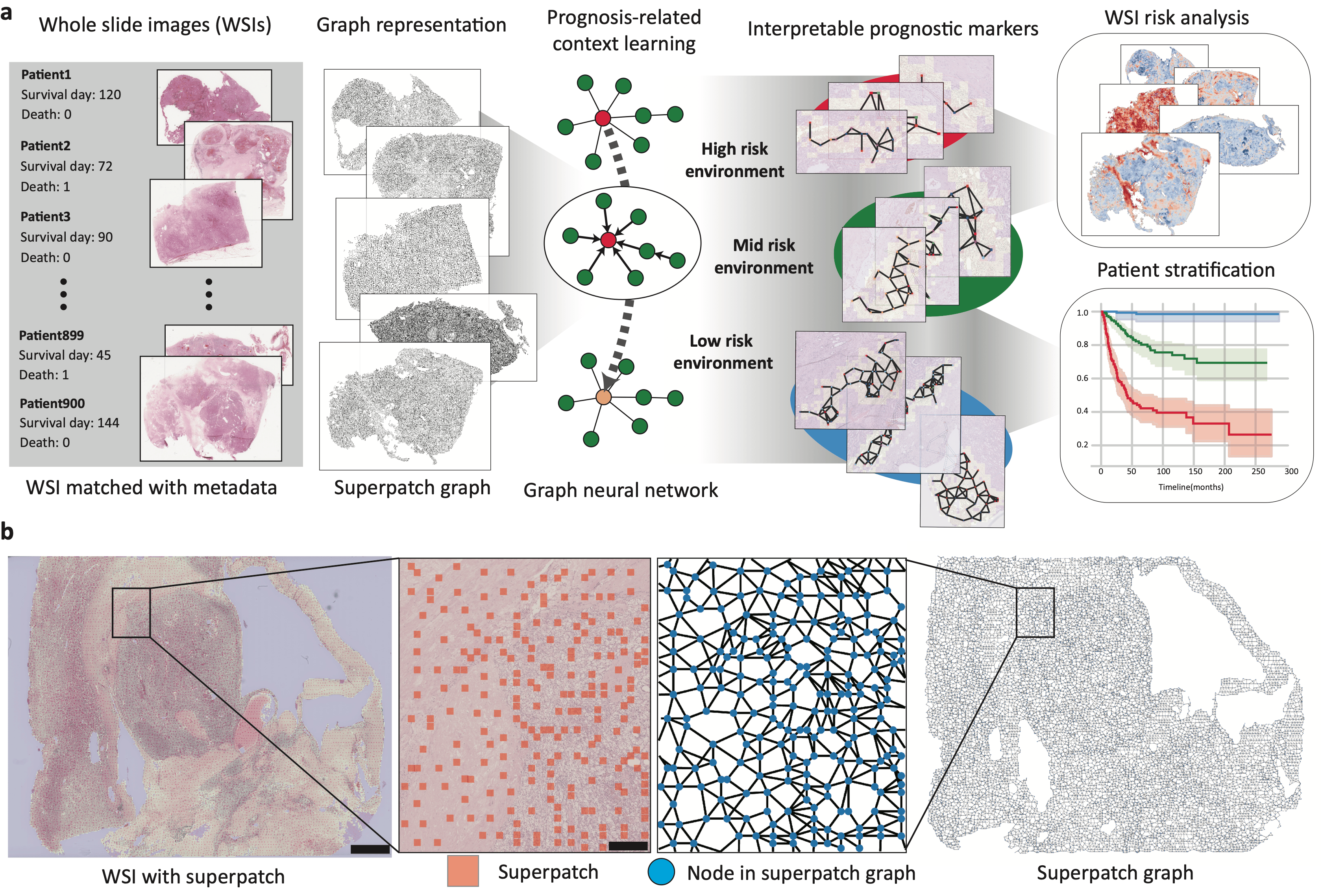
At first, we tried to simply use the GNN for contextual feature analysis, but several challenges exist that must be carefully dealt with to detect the contextual features using the WSIs. The first challenge is the use of WSI, which is a gigapixel image that brings computational burdensome. Another challenge is to reflect the physical interaction of each cell in TME using GNN. As the solution, we present the WSI as a compressed network of superpatch. We suggest the superpatch which is the aggregation of small patches with similar pathological features. On the other hand, we propose and demonstrate a position-aware GNN that incorporated the physical location of each node to better representation of cellular interactions in TME.
As the demonstration, we used the TEA-graph to predict the prognosis of cancer patients and TEA-graph achieved pathologist-level accuracy in predicting the prognosis of cancer patients. Also, we introduce the interpretation framework based on the integrated gradient (IG) method that quantifies the influence of each pathological feature on the prognostic results of TEA-graph. The interpretation method changed the black box like GNN into understandable contextual pathological markers which is important in the biomedical field. We could find the unfavorable contextual features related to the coexistence of angiogenic features with the tumor cell, and immune cells named the active granulation and angiogenic focus (AGAF).
Approximately three years ago, we discussed developing a simple convolutional neural network-based model to classify the subtypes of cancer. However, the active collaboration of engineers and pathologists lead the project to have both technical and clinical meaning. In the future, we demonstrate the clinical utility of the biomarkers extracted by the interpretation module to better show the value of TEA-graph. We hope that this study will facilitate further biomedical research using deep learning.
Follow the Topic
-
Nature Biomedical Engineering
This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.

Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Medical Ultrasound: Emerging Techniques and Applications
Publishing Model: Hybrid
Deadline: Jan 29, 2027




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in