High fiber content degradable biocomposites using green modular polymerization
Published in Materials
Wood fibers are excellent reinforcement candidates for biocomposites, but it is hard to achieve high fiber content. The primary motivation for using wood fibers is their renewable resource, wide availability, high mechanical properties, and low density. So far, a lot of effort has been made to develop high-fiber thermoplastic composites through melt processing methods like compounding. However, the compatibility of fibers has been an issue since most commercial thermoplastic polymers are hydrophobic, unlike wood fibers. This caused dispersion issues, especially at large reinforcement levels. Consequently, the high viscosity, mechanical fiber damage, and agglomeration have prevented the development of high fiber content biocomposites of high mechanical properties.
Motivation. Although natural fiber biocomposites have a biobased origin, they seldom contribute to sustainable development, considering all processes involved in their life cycle. The key is to look at the entire cycle, meaning that the biobased origin must be linked to environmentally friendly processing, production must be energy efficient, and the final material must be degradable or recyclable. In this sense, in-situ matrix formation within the fiber network seems to be the logical and energy-efficient strategy.
The challenge. We started looking at ring-opening polymerization of e-caprolactone (CL) as the model system to build knowledge around in-situ polymerization challenges. CL was chosen because of its thermodynamically favored polymerization features, ability to be polymerized by a wide range of catalysts, and ability to react with cellulose reinforcement for improved interfacial adhesion. The final polymer, poly(e-caprolactone) (PCL), is hydrolyzable and has a low melting temperature that enables thermoforming. However, the synthetic challenge is that water initiates polymerization. In ambient conditions, a typical cellulosic fiber has at least 5 wt% moisture content. During in-situ polymerization, residual moisture significantly reduces the final polymer molecular weight, where the reduction is a function of the fiber content, Fig. 1 shows this trend.
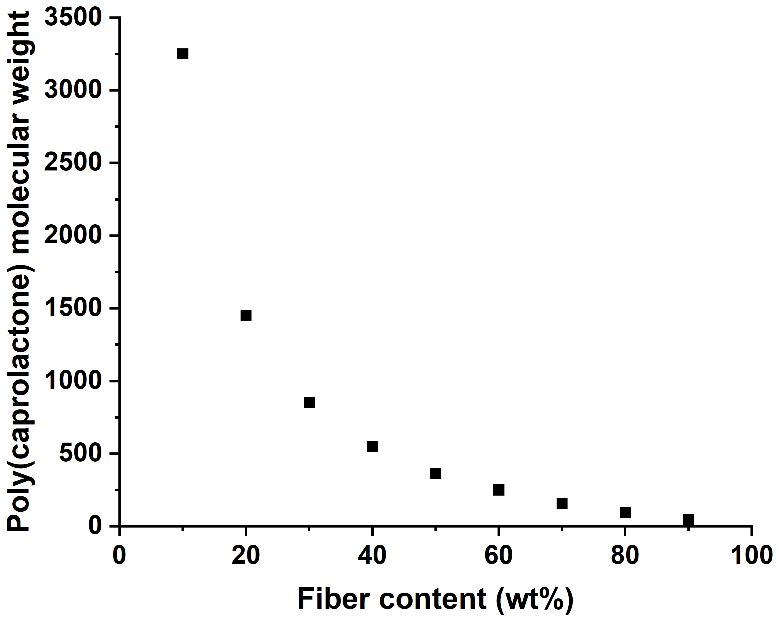 Fig. 1 Theoretical challenge of forming poly(e-caprolactone) via in-situ polymerization in a fiber network (with 5wt% moisture).
Fig. 1 Theoretical challenge of forming poly(e-caprolactone) via in-situ polymerization in a fiber network (with 5wt% moisture).
In-situ chemical drying. Extensive system drying (under inert gas flow, extremely high vacuum, and/or high temperatures) can eliminate moisture. Nevertheless, it is energy-intensive, difficult to scale up, and may cause irreversible hydrogen bonding (hornification) of the fibers. We initially started exploring different methods of in-situ chemical drying to enable a dry system while simultaneously avoiding hornification. In this strategy, after vacuum filtration of cellulosic water suspension, the network goes through several solvent exchange procedures (to reduce the water content), followed by a final chemical reaction that consumes the water molecules. We started polymerizing the degradable polymer matrix in-situ when the system was dry, in a solvent-swelled fiber network system. We explored many methods, such as copper sulfate in acetic acid, acetic anhydride, and cyclic anhydrides. For the polymerization, we explored numerous different catalytic systems such as stannous octoate, diphenyl phosphate, and methanesulfonic acid in solvent systems including acetone, toluene, acetic acid, tetrahydrofuran, acetonitrile, propylene carbonate, and dioxane. Additionally, we tried running the polymerization at higher temperatures (helpful for water evaporation). These attempts concluded that acetic anhydride chemical drying, followed by ring opening polymerization of CL within fiber networks soaked in toluene at 110°C, was sometimes successful. The problem was that the moisture content in the fibers changed on a daily basis and depended on the fiber content of the system, necessitating a new optimization for each sample.
Modular green polymerization. To overcome the reproducibility problems, we applied a new green in-situ polymerization strategy for the in-situ polymerization of the degradable matrix via the condensation polymerization of caprolactone oligomers. The ε-caprolactone oligomers were synthesized through ring-opening polymerization of ε-caprolactone with glycerol as an initiator, catalyzed with methane sulfonic acid, and partially end-capped with succinic anhydride. The following oligomers were polymerized at 140°C with stannous octoate as the catalyst. The high temperature removes water molecules from the system to yield relatively high molecular weight polymers. Note that in-situ polymerization of ε-caprolactone monomers instead of oligomers is not feasible as the final polymer yield is low due to the higher evaporation rate. We screened a series of oligomers with different molecular weights and end group rations. We found that functionally balanced three-arm caprolactone oligomers with number average molecular weight of 1500 g/mol lead to the formation of thermoformable polymeric films with ~40 wt% gel (crosslinked) and ~60 wt% of polymer with an average molecular weight of ~10 000 suitable for the biocomposite matrix. For the biocomposites, wet fiber networks were solvent exchanged for acetone, and the oligomers infiltrated into the fiber networks. After infiltration, the polymerization was performed at 140°C for 14 h in the network, followed by densification (Fig. 2). As a result of this strategy, we circumvent the moisture problem and achieve a reproducible polymerization system for different fiber contents and reinforcement types.
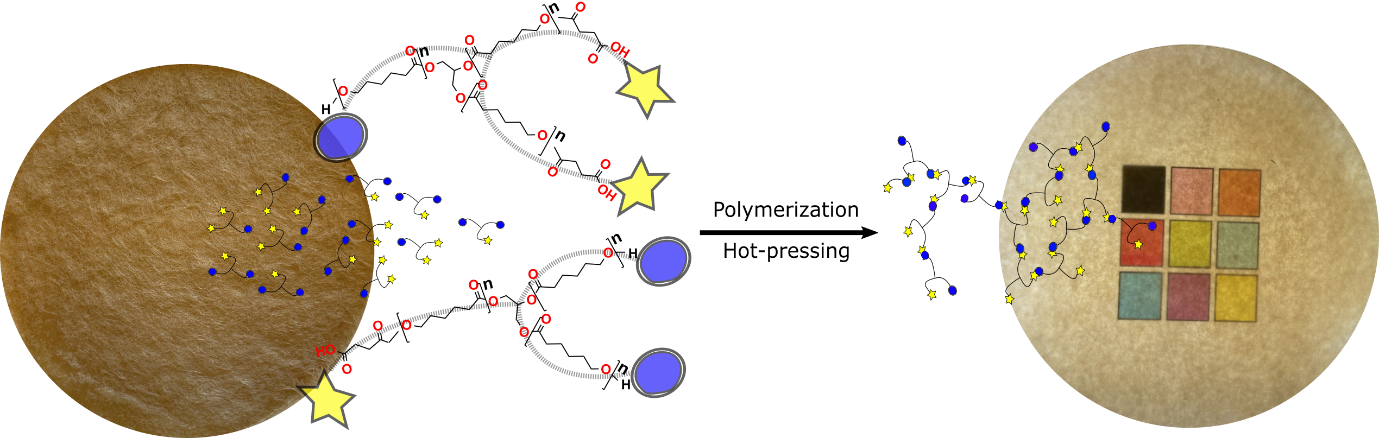 Fig. 2 Polymerization of modular and degradable units within a pre-formed fiber network, followed by densification.
Fig. 2 Polymerization of modular and degradable units within a pre-formed fiber network, followed by densification.
The potential of the strategy. This modular green polymerization concept allowed us to make biocomposites of very high fiber content (>50 wt%), while retaining adequate dispersion, resulting in mechanical properties significantly superior to literature equivalent materials (Fig. 3a). Achieving good dispersion at high fiber content is not feasible with regular processing techniques such as melt compounding.
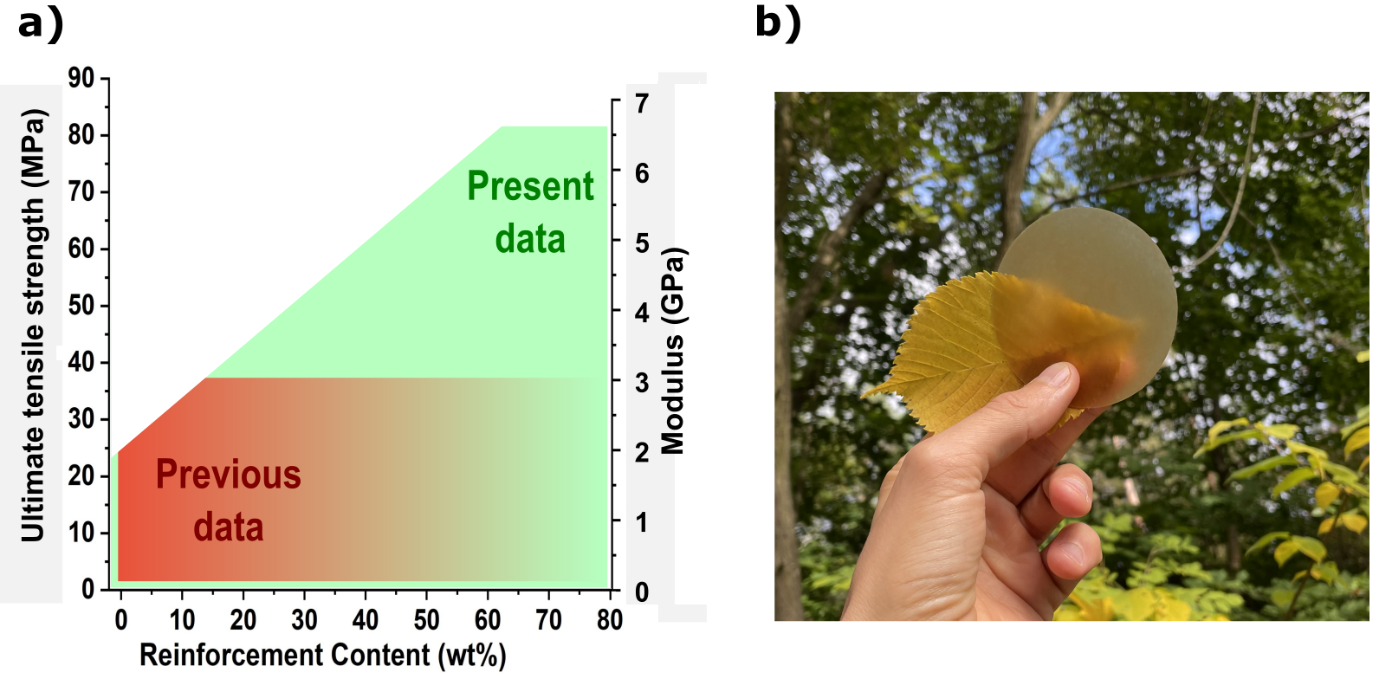 Fig. 3 a) The potential to create stronger biocomposites through well-dispersed high fiber content (data related to PCL cellulose reinforced biocomposites). b) A photograph of PCL/25% wood fiber biocomposite.
Fig. 3 a) The potential to create stronger biocomposites through well-dispersed high fiber content (data related to PCL cellulose reinforced biocomposites). b) A photograph of PCL/25% wood fiber biocomposite.
This strategy also allows for a more accurate assessment of the influence of reinforcement types (fibers and fibrils) on mechanical properties, as reinforcement damage and agglomeration issues are minimized. We made a series of biocomposites with different reinforcement content using microfibrillated cellulose or wood fiber to investigate the effects of reinforcement geometry on mechanical properties. Other than mechanical properties, we showed that the formed matrix's hydrolytic degradation rate also depends upon the reinforcement geometry. Therefore, the mechanical properties, ductility, and degradation rate can be altered depending on the reinforcement. Using this method, we produced thermoformable, translucent, and high-performance polycaprolactone-based biocomposites with customizable degradation rates (Fig. 3b). In summation, this research is in the context of sustainable development of biocomposites through the development of a novel green in-situ polymerization strategy suitable for high fiber content and degradable/recyclable biocomposites. This reactive process brings minor damage to the reinforcement and allows for good dispersion, resulting in considerably higher mechanical properties than similar biocomposites. Additionally, being capable of scaling up and performing solvent-free at elevated temperatures.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in