

The paper "Defect-induced tuning of polarity-dependent adsorption in hydrophobic-hydrophilic UiO-66" has been published in Communications Chemistry, 5, 120 (2022).
The link can be found here: https://doi.org/10.1038/s42004-022-00742-z.
Metal-organic frameworks (MOFs) are an extensive class of crystalline, porous hybrid materials consisting of secondary building units (SBUs), like metal ions or metal-oxide clusters, connected by organic ligands. For years, scientists have been striving to synthesize new materials, creating new combinations of ligands and metals, for many applications such as adsorption, separation, conductivity, or magnetism. The less studied side of porous materials is their modification, e.g., by introducing defects and thus changing not only their properties but also their chemical nature. This can be called refinement, although not always so easily controlled. Perhaps that is why the modification is less desirable than the creation of new ones. An example of a material that changes its character dramatically through modification is zirconium-based UiO-66. The control of vacancy introduction depends largely on the synthesis temperature: the lower, the more "leaky" the structure. Along with defects, the hydrophilicity of the material changes, and hence its sorption properties and capacity. However, understanding the mechanism of this two-dimensional change (i.e. polarity of the adsorbent and adsorbate) is not trivial, and using experimental methods alone: is almost impossible. At that point, like a knight on a white horse, molecular simulations enter.
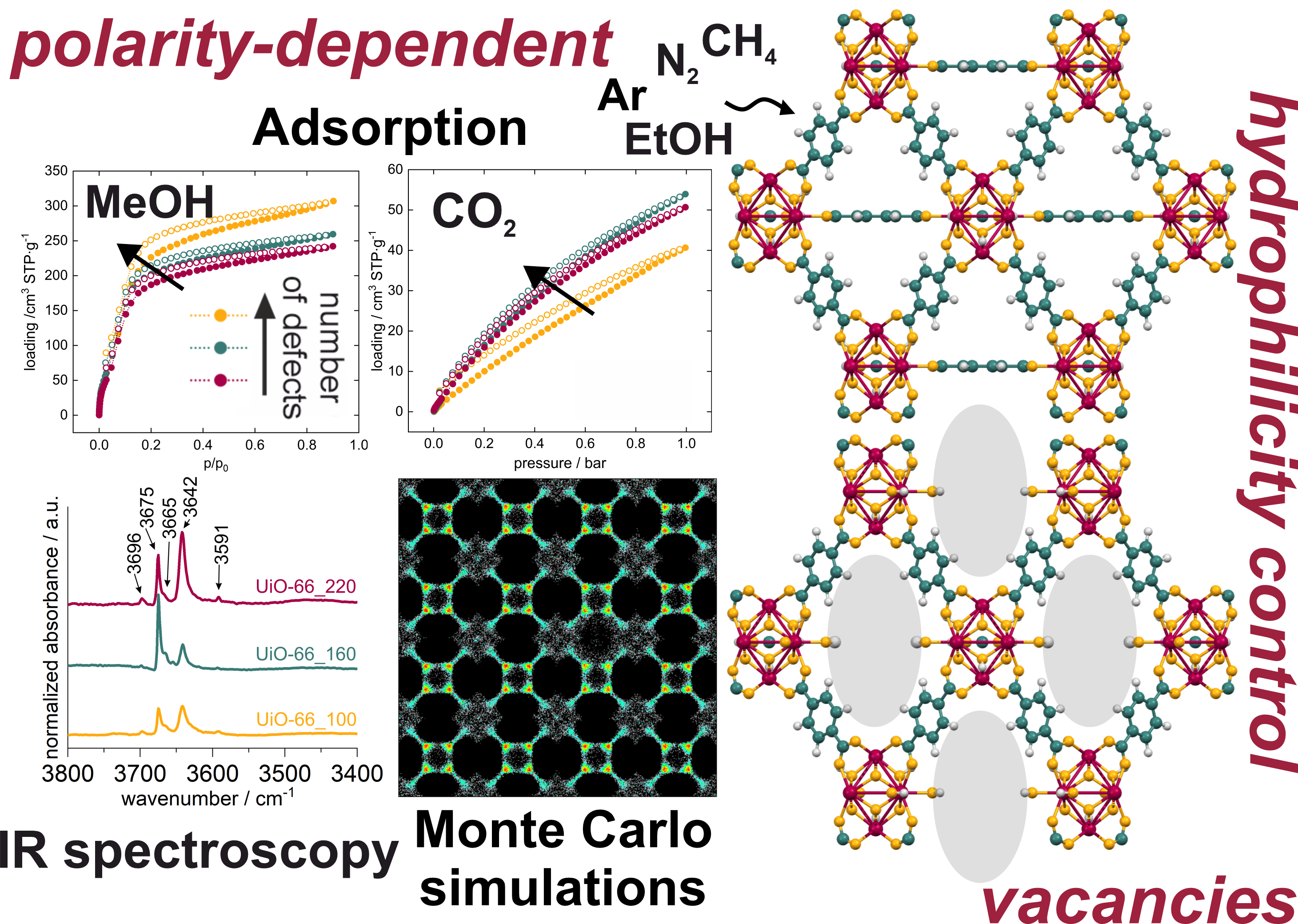
Many researchers from various fields of science use molecular simulation as an important tool for understanding basic molecular theories. Computational methods, in general, can be performed at two levels: quantum (e.g. Density Functional Theory, DFT) and classical (e.g. Monte Carlo, MC). Moreover, the basic idea of the MC calculations is to estimate the mean value of the system by sampling a set of configurations with an appropriately given probability of trial moves. The former treats electrons as the basic interactive particles of the system, while the latter sees whole atoms or groups of atoms as fundamental individuals as interacting ‘particles’ of the system. However, confidence in the predictions of molecular simulations depends on the reliability and accuracy of the selected force fields. Unfortunately, quantifying the uncertainties related to the form of force fields and their parameters is also not straightforward, but is a fundamental part of molecular modeling.
In this work, we used a previously developed forcefield for water adsorption in UiO-66 to fully understand the mechanism of the adsorption of polar and nonpolar molecules in more hydrophobic or more hydrophilic material. Using a variety of methods, from volumetric adsorption experiments and IR spectroscopy measurements to DFT+D and Monte Carlo computational methods, it was possible to track a two-dimensional change in polarity.
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Jun 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: May 30, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in