Integrated transcriptome study of the tumor microenvironment for treatment response prediction in male predominant hypopharyngeal carcinoma
Published in Cancer

Hypopharyngeal carcinoma (HPC), a predominantly male cancer1, occurs at hidden sites and does not cause obviously uncomfortable symptoms at early stage, both of which lead to the diagnosis of HPC patients mainly at advanced stage and the worst prognosis among all head and neck squamous cell carcinoma2. In present clinical practice, doctors apply TPF (taxol, cisplatin, 5-FU) induction chemotherapy plus cetuximab, the combined treatment, for advanced HPC patients to reduce tumor volume and then re-evaluate the possibility of radically surgical resection of the tumor. However, the objective response rate to the combined treatment is only about 50%3, and no method is available to evaluate the curative effect before the combined treatment based on CT scanning or reliable biomarkers. The conditions of treatment-resistant patients are deteriorated, and they miss the best time window for surgery or other possible therapies, suffering financial and psychological stress.
Inspired by researches from other cancers such as melanoma and lung adenocarcinoma4,5, we believe that a thorough understanding of the tumor microenvironment (TME) in HPC will promote the identification of accurate and effective biomarkers assisting in the stratification of patients resistant to the combined treatment. With the aim to depict the multicellular ecosystem of HPC, we collected clinical HPC tumor samples from different treatment timepoints with different treatment efficacy for integrated transcriptome analyses. All the work was conducted by two groups from Beijing Tongren Hospital and Tsinghua University, focusing on clinical research work of head and neck cancer and synthetic and system biology research respectively. Zhigang Huang and Zhen Xie conceived this project and provided guidance and supervision for the successful implementation. As the chief physician of Otorhinolaryngology Head and Neck Surgery, Yang Zhang established the standards of collecting clinical tumor samples to ensure the access of various cell types and gathered all needed samples for our study. Along with the efforts from Wei Guo, Gaofei Yin, Wen Gao and Lifei Feng, we successfully obtained ethical approvals and well-preserved clinical samples for the HPC cohort, which were important for treatment response prediction explorations. Additionally, Dr. Gan Liu established the sample preparation protocol for single-cell RNA sequencing analysis, and smoothed the communications between two groups with his multidisciplinary academic backgrounds. PhD student Minzhen Tao learned various scRNA-seq analysis technics from scratch and conducted all the bioinformatic analyses to deduce the meaningful findings in this study, with useful suggestions provided from Hui Ning and Jin Gu in the process of SVM model construction and data preprocessing.
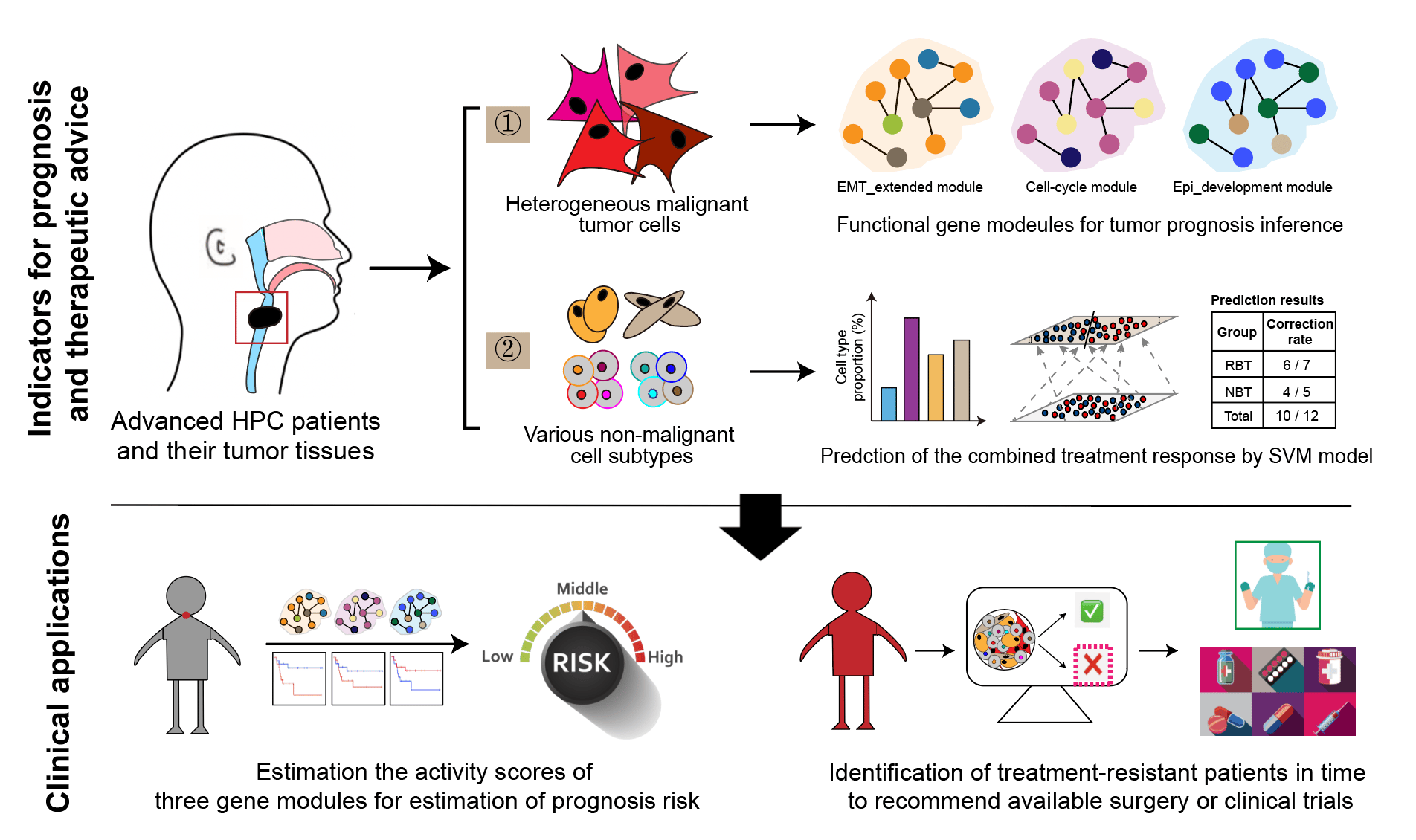
In the study, we performed single-cell RNA sequencing (scRNA-seq) on 15 clinical HPC tumor samples from 8 participants, combined with transcriptome expressions of a HPC cohort with bulk RNA-seq. Through the high-resolution profiles generated from scRNA-seq, we identified various cell types, including malignant epithelial cells, endothelial cells, fibroblast cells, B cells, myeloid cells, T and NK cells, all of which were also confirmed by multiplex immunohistochemistry with specific antibodies. Therefore, we characterized the major difference between malignant and non-malignant cells in downstream analyses.
On the one hand, we applied non-negative matrix factorization as well as related correlation analyses on malignant tumor cells, from which we identified 5 functional biological gene modules from heterogenous tumor cells. Comparing the transcriptional expression differences of the modules among sample groups, we revealed that high activities of cell-cycle and EMT_extended gene modules and low expression of Epi-development gene module would lead to the resistance before the combined treatment. Furthermore, survival analyses suggested that three gene modules could be used for prognosis prediction in advanced HPC. On the other hand, through systematic biomarker and gene pathway characterizations, we identified cell subtypes in other non-malignant environmental cell types, which could be further classified into anti-tumor and pro-tumor subtypes based on their biological functions. When analyzing the differences between groups in terms of cell-subtype proportions and intercellular interactions, we found that the treatment-resistant samples collected before the treatment showed stronger immunosuppressive, pro-angiogenic, and extracellular matrix modeling functions.
Then, taking advantage of the convenience of bulk RNA-seq and the high resolution of scRNA-seq, we developed a model to predict patient’s response to the combined treatment based on non-malignant cell compositions in TME. With a signature gene matrix derived from 15 well-characterized subtypes and gene expression data of the HPC cohort, we digitally estimated the non-malignant cell abundances via CIBERSORTx6, which were further linked to the treatment responses by training a non-linear support vector machine (SVM) binary classifier model. The model had a relatively high prediction accuracy, with the overall correction rate at about 83% in a small-scale prospective trial with additional 12 treatment-naïve samples, showing the great potential value for future clinical applications.
In summary, for the first time, we provided a comprehensive and unique resource revealing the landscape of HPC TME at single-cell resolution, uncovered three functional gene modules of malignant tumor cells associated with prognosis, and established a quantitative classifier model between non-malignant subtypes’ composition and patients’ responses to the combined treatment. Especially, the treatment response prediction model used only bulk RNA-seq and will be convenient and economic to provide therapeutical advice in future clinical practice, enlarging the possibility of laryngeal function preservation and providing a better therapeutic experience for advanced male predominate HPC patients.

Figure. Graphical summary of our integrated transcriptome study in male predominant hypopharyngeal carcinoma
Reference:
- Bradley PJ. Epidemiology of Hypopharyngeal Cancer. Adv Otorhinolaryngol 83, 1-14 (2019).
- Garneau JC, Bakst RL, Miles BA. Hypopharyngeal cancer: A state of the art review. Oral Oncol 86, 244-250 (2018).
- Huang TQ, et al. Induction chemotherapy for the individualised treatment of hypopharyngeal carcinoma with cervical oesophageal invasion: a retrospective cohort study. World J Surg Oncol 18, 330 (2020).
- Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189-196 (2016).
- Lambrechts D, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24, 1277-1289 (2018).
- Newman AM, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773-782 (2019).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in