Ionic Interactions Utilized in Covalent Organic Frameworks
What Makes Covalent Organic Frameworks Interesting?
Covalent organic frameworks (COFs) are crystalline materials constructed from organic building blocks with intrinsic porosity. Since their discovery in 2005, they have received increasing attention from various research fields, emerging as a highly tunable and versatile platform. This is due to the vast number of building blocks available, which can be tailored to specific functionalities and induce desired properties. Additionally, the reticulation of the building blocks can be achieved using a multitude of organic linkages or nodes, which allows for further diversification.
Structurally, COFs can be divided into one- (1D), two- (2D), and three-dimensional (3D) frameworks. Usually, the topology of the framework can be targeted ab initio as it depends on the geometry and connectivity of the respective building units used. 2D COFs have layers made from covalent bonds that typically stack through non-covalent interactions such as pi–pi stacking and/or hydrogen bonding, which directly affect the crystallinity and porosity of the material. However, this targeted approach does not always work, and sometimes outstanding structures are obtained that were not anticipated.
The Motivation
Our motivation arose from a work published by our group in 2018.[1] An aluminate-based MOF (Al-Td-MOF-1) was synthesized from lithium aluminum hydride. Here, tetrahedral anionic aluminate ([AlO4]−) units are connected by linear dihydroxy-functionalized linkers. If we simplify the building units into geometric shapes, the aluminate is equal to a tetrahedron, and 1,4-dihydroxbenzene equal to a linear stick. The combination of these should result in the topology of a three-dimensional diamond (dia) based net, which indeed was found to be the case for Al-Td-MOF-1. However, a large tetragonal distortion was found, which was not anticipated. The distortion was discovered to result from electrostatic interactions between the lithium cation and the [AlO4]- units, allowing for efficient charge compensation.
Inspired by this work, we wondered: Is it possible to synthesize a metal-free analogue of Al-Td-MOF-1?

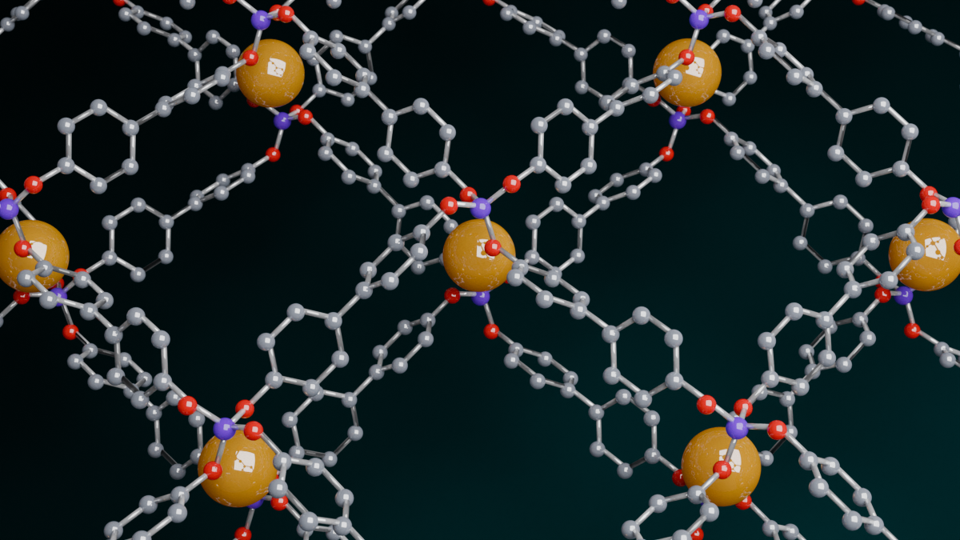
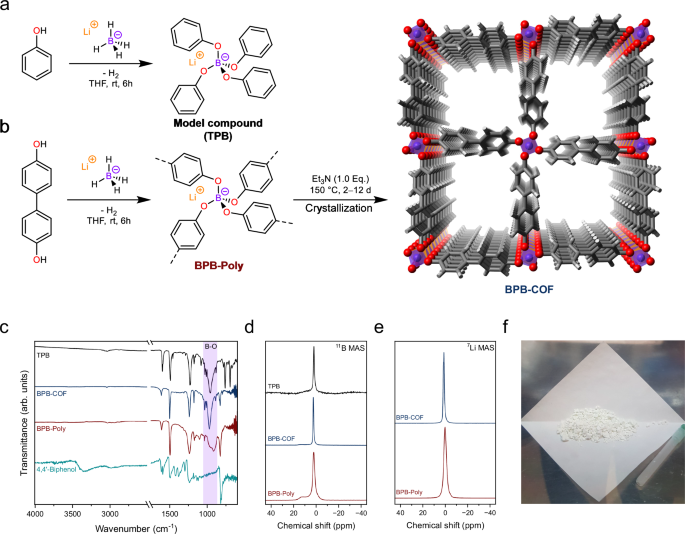
To synthesize a metal-free analogue, we aimed to replace the aluminate for a tetratopic borate ([BO4]-) node due to the similarities, as both are anionic tetrahedral motifs. Indeed, by substituting lithium aluminum hydride with lithium borohydride and 1,4-dihydroxybezene for 4,4'-dihydroxybiphenyl we were able to synthesize a borate-based covalent organic framework (BPB-COF). BPB-COF was found to crystallize with a morphology of beautiful hollow spheres made from 100-200 nm long agglomerated crystallites.
With the compositional analysis confirming the integrity of our material, the remaining question was how the crystal structure of BPB-COF looks. Is it similar to Al-Td-MOF-1?

Solving a Puzzle Without Reference
Even though there were many different struggles regarding this work (a major water damage at the institute leading to critical instruments being out of order for multiple months, pursuing a synthetic dead end for 6 months, etc.), developing a structural model turned out to be the biggest challenge for this work.
As we obtained a polycrystalline material, our only option for a structure solution was to rely on powder x-ray diffraction (PXRD) and simulate a fitting structural model, which we then refined using experimental PXRD data.
To solve the crystal structure, we first started by indexing possible unit cell parameters from the PXRD pattern. This gave us a selection of three suitable tetragonal unit cells. Similar to Al-Td-MOF-1, we have a tetrahedral borate node connected by a linear linker, leading us to believe that the resulting topology should be a dia-net with the possibility of a tetragonal distortion. Additionally, it should be mentioned that COFs with dia topology are interpenetrated to some degree, which further increases the number of possible structural models. Over the span of many months and hundreds of hours, we simulated different dia-nets within the three proposed unit cells that were obtained through indexing. However, no matter what structural model we developed, one characteristic diffraction was always absent. We started to investigate other three-dimensional topologies, but this was also unsuccessful. With no topology seemingly fitting the PXRD pattern, we started to disregard any topology and iteratively adjusted the position of the linker within the unit cells. We observed that when the linker is located in-plane, the missing diffraction would occur. This was very unexpected, as an in-plane arrangement of the linker would not result in a 3D structure but a 2D structure like the square lattice (sql) topology.
This finding however, only led to more confusion as:
- 2D structures utilize "flat" in-plane oriented building units, while the [BO4]− unit is tetrahedral - how can a tetrahedral building block result in a 2D structure?
- In this structural model, this would result in stacked anionic nodes, which should be unfavoured due to electrostatic repulsion.

Was this just a fluke, or is there any structural feasibility?
We realized that, although biphenyl is a linear linker, the angled B–O–C-linkage and the flexibility around the borate unit enable the reticulation into a 2D layer. While this resolves the first point, it does not address the second point, which left us wondering: How would the individual layers stack?
Looking for possible answers, I remembered two articles I had read on molecular tetraphenoxy borates where they also discussed their crystal structure.[2,3] I re-read the articles in more detail and saw that the counterion is tetrahedrally coordinated by two phenoxy atoms of the ligands and two solvent molecules. In the second article, it was shown that even pillaring of molecular borates was facilitated by the counterion, leading to the formation of 1D chains.
What if the counterion is not located within the pores coordinated by solvents but rather fixed in the interlayer space, tetrahedrally coordinated by two phenoxy moieties of adjacent layers?
Further pursuing this idea and developing a structural model, the complementary lithium counterions were added in the interlayer space in between adjacent borate linkage sites, resulting in LiO4 tetrahedra. This model justifies the stacking of the anionic linkages as it allows for efficient charge compensation. Furthermore, the unit cell parameter obtained was c = 5.01 Å, which explains the larger layer-to-layer distance compared to COFs stacked by pi–pi interactions or hydrogen bonding, which typically reach up to ~3 Å.

This finding demonstrates that like Al-Td-MOF-1, the counterion is structurally and topologically significant. However, here not only a distortion of the expected topology occurs, but even a completely different topology is enabled, resulting in a 2D instead of a 3D structure.
2D or not 2D - That is the Question
Speaking of dimensionality, we were not quite sure how this structure would be best described. So far, the counterion was always disregarded for charged COFs, but doing so here would neglect the critical structural role of the counterion. On the other hand, trying to include it would make the [BO4]− unit a 6-coordinated building block, which is very unintuitive and would result in a 3D topology. We decided that since for covalent organic frameworks the focus relies on covalent bonds, the lithium ion should not be considered topologically, as they are not covalently bound. However, as a compromise, we decided to highlight the structural importance of the lithium ion by coining it a "pillared" instead of stacked 2D COF, finally being described as a 2D sql-net pillared by interlayer lithium ions.
Challenging our Simulation
Even though the Rietveld refined model provided good Li–O and B–O-bond distances and was in accordance with the experimental data, we were still skeptical about our structural model and decided to test whether we could demonstrate any structure-function relationship. We reasoned that since the layers are made from more robust covalent bonds, it should be possible to selectively target the weaker ionic interactions pillaring the layers.

Indeed, we were able to confirm our theory by simply using a polar solvent (methanol) that would interact with the lithium ions, disturbing the ionic [BO4]− – Li+ interactions, resulting in the exfoliation into ultrathin layers. The obtained nanosheets were characterized using transmission electron microscopy (TEM) and atomic force microscopy (AFM), showing exfoliated layers with a thickness below 1 nm, demonstrating that the ionic interactions pillaring the layers can be selectively targeted to give ultrathin covalently bonded nanosheets and further validating the structural model.

Now, the article is out and we hope that the structural elucidation of BPB-COF could demonstrate how important non-covalent interactions can be - even for covalent organic frameworks.
[1] Angew. Chem. Int. Ed. 2018, 57, 16683.
[2] Eur. J. Inorg. Chem., 2006, 1690-1697.
[3] J. Chem. Soc., Dalton Trans., 2000, 3100-3105.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in