MetaMARC: Using Machine Learning to Identify Antimicrobial Resistance
Published in Microbiology

Antimicrobial resistance (AMR) refers to the ability of a micro-organism to resist the effects of an antimicrobial drug. AMR is an increasingly important public health issue because it renders standard treatments for bacterial infections (e.g., antibiotics) ineffective, leading to longer and more complicated infections. Approximately 2 Million people each year in the United States contract a resistant infection and other 20,000 people die of resistant infections. AMR can occur when bacterial chromosomes and/or plasmids acquire genes that help the bacterium to evade the antibiotic or make it ineffective; these are referred to as AMR genes.Because bacteria can exchange DNA, it is important to understand the entire genetic repertoire of AMR genes within a given microbiome. This is termed the resistome, and refers to the set of all AMR genes found in pathogenic and non-pathogenic bacteria; it defines the potential resistance of a given microbial community to all known antibiotics. Previously, analysis of AMR relied on culture-based studies that involve isolating specific bacteria from a sample and then observing the bacteria’s ability to grow in the presence of different types of antibiotics at differing concentrations. Although these culture-based methods can be quite effective and reliable, they are limited in the number and type of bacteria that can be studied. One promising avenue to study the resistome in a clinical or environmental sample, in a manner that reduces these limitations, is to directly sequence the genome of all bacteria. A technique called shotgun metagenomics can sequence all the DNA of a sample yielding millions of gene fragments —these data can be used to identify the AMR genes without the need to culture the sample for specific bacteria.
Since metagenomics can be effective for identifying the AMR genes in a sample, many projects have generated shotgun metagenomic data to characterize the resistome across various time periods and settings, including clinical facilities, public facilities, and food production chains. These datasets often necessitate analysis of an extremely large number of samples. Given all the current and forthcoming data, it is important that methods for analyzing the resistome are scalable, i.e. they can be applied to increasingly large data sets without compromising the quick turnaround of results.
Previously, our lab presented AMRPlusPlus which maps all shotgun metagenomics sequences from a given sample to the MEGARes database using a fast aligner, and then reports all AMR genes which have 80% of their reference sequence covered by the sequence data. Although this method is effective in finding AMR genes in a metagenomic sample, it relies heavily on alignment and is unable to predict AMR genes that do not have strict sequence homology to the database reference, e.g., potentially new resistance genes for existing or novel antibiotics. Thus, in order to increase the accuracy of the prediction, we knew we would have to develop a more flexible model to classify the reads. For this reason, we developed Meta-MARC, which uses a statistical technique called Hidden Markov Models (HMMs) to classify shotgun metagenomic reads, thus predicting AMR genes more accurately.
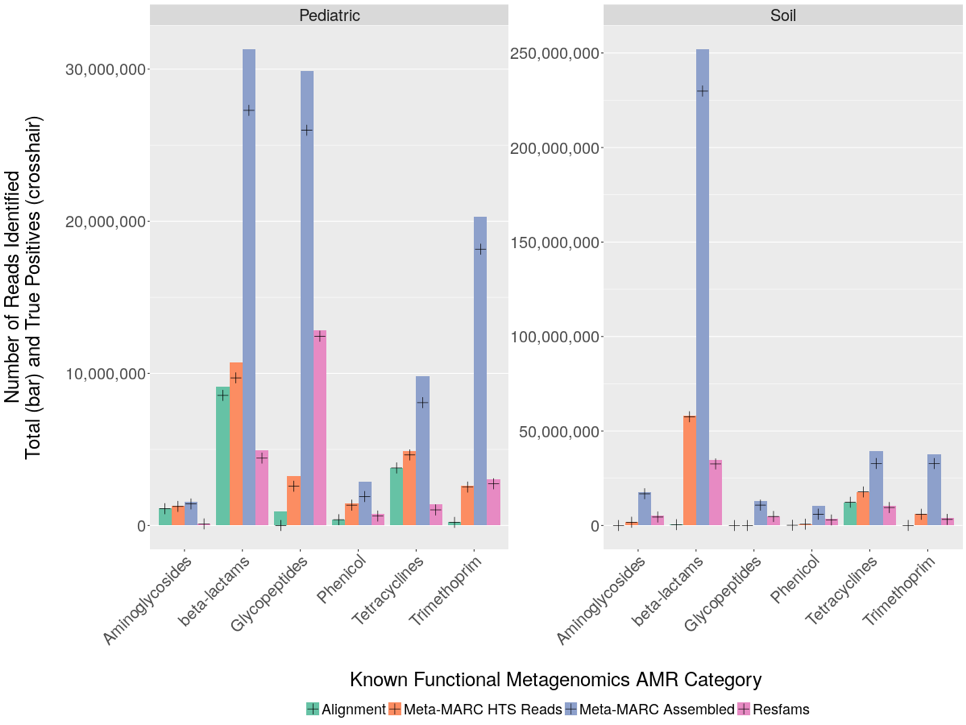
We tested Meta-MARC on simulated and real metagenomic data. Through these experiments we were able to demonstrate that Meta-MARC has significantly higher sensitivity in comparison to competing methods. This sensitivity allows for the detection of sequences that diverge from reference sequences of known AMR genes. As shown in the above figure, we demonstrate that Meta-MARC is able to identify a significantly larger number of sequence reads from almost all resistance classes in both human- and soil-derived samples. This functionality is imperative to expanding existing databases of known antimicrobial resistance genes.
See the full paper in Communications Biology here:
https://www.nature.com/articles/s42003-019-0545-9
The source code for Meta-MARC is available here:
https://github.com/lakinsm/meta-marc.
Dr. Mattia Prosperi at the University of Florida and Dr. Noelle Noyes at the University of Minnesota contributed to the preparation of this post.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
DNA repair and human disease
Publishing Model: Hybrid
Deadline: Oct 31, 2026
Cell death and inflammatory signalling
Publishing Model: Hybrid
Deadline: Oct 28, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in