Modeling stem cell fate dynamics
Published in Bioengineering & Biotechnology
What is a cell type? Is it different from a cell state? How do we define biological systems that are constantly in flux? These are some of the questions that reverberate within me when I think about stem cell engineering. After years of grappling with the inherent complexities of stem cell systems – everything from cell-cell heterogeneity to phenotypic dynamics – my engineering proclivities drove me to systems biology as a quantitative solution to holistically describe, and maybe even predict, stem cell differentiation.
In 2014, as a postdoc in George Daley’s lab at Boston Children’s Hospital, I was swept up in the excitement surrounding a new computational algorithm – CellNet – which had recently been pioneered by lab members Patrick Cahan and Samantha Morris. CellNet [1,2] promised to revolutionize cell engineering by giving us a benchmark – a single, quantitative metric – to judge how closely our cells resembled bona fide human cells. Then, in lieu of blindly screening thousands of molecules, we could instead use the algorithm to accurately predict molecular tweaks to increase parity even further. Seeing the outputs of an algorithm translate into successfully reprogramed cells inspired me to explore how this could be applied to probe the complexities of stem cells. To extend beyond CellNet’s original 16 discrete, terminal cell types (e.g. heart, lung, skin, etc.), I began thinking about how we could use the algorithm to describe the more nuanced, intermediate stages of stem cell differentiation.
As a bioengineer, I saw the hematopoietic hierarchy as the perfect system to model; the differentiation paths and all of the intermediate stages of blood cells arising from the hematopoietic stem cell were precisely defined. From George’s perspective, there were still several blood cell types that we had yet to faithfully recapitulate in culture. We wondered: would it be possible to reconstruct the hematopoietic hierarchy via a modified “BloodNet” algorithm? Would it teach us how to better differentiate stem cells along specific blood lineages? So, I – a bioengineer – jumped into the deep end of hematology.
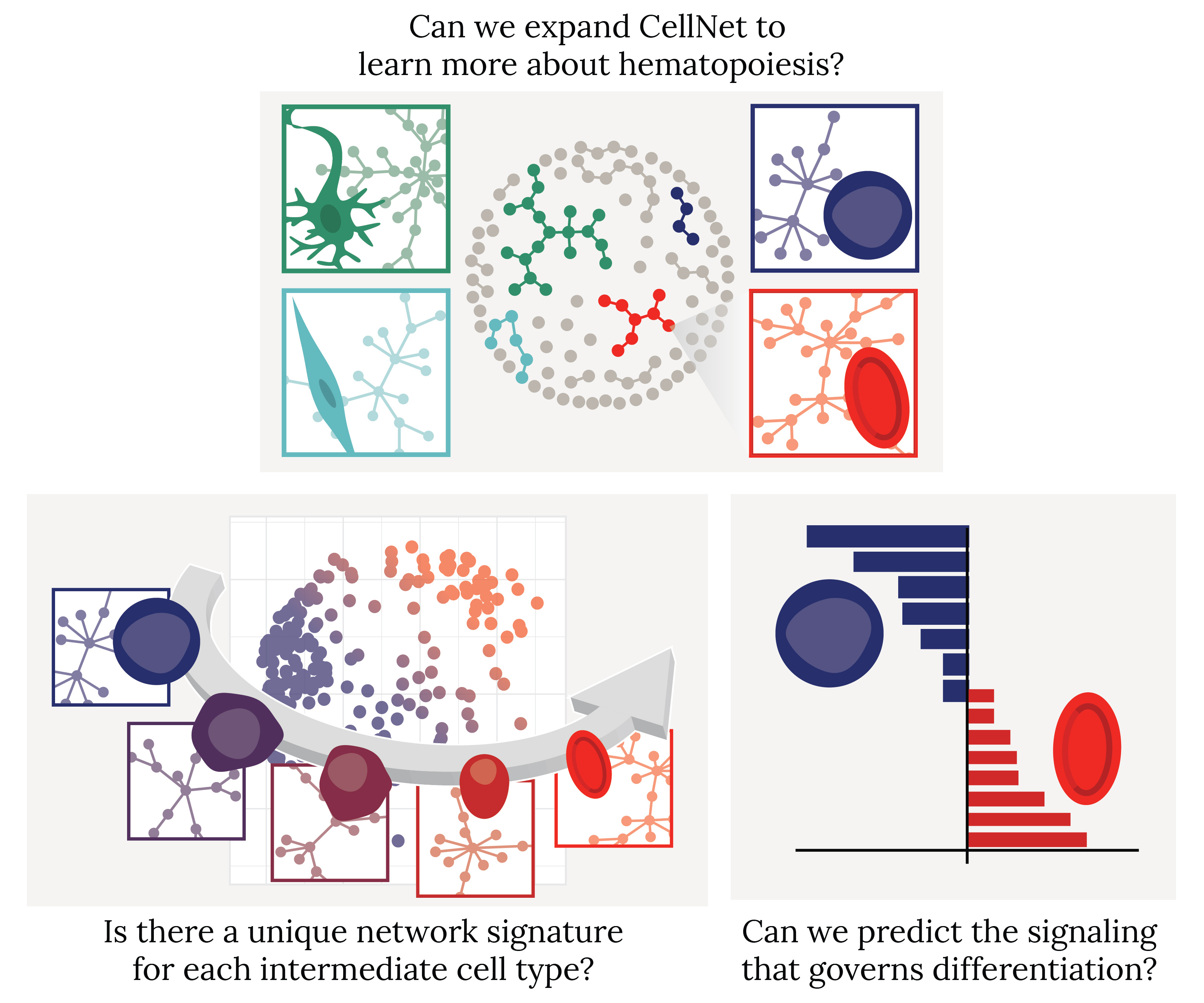
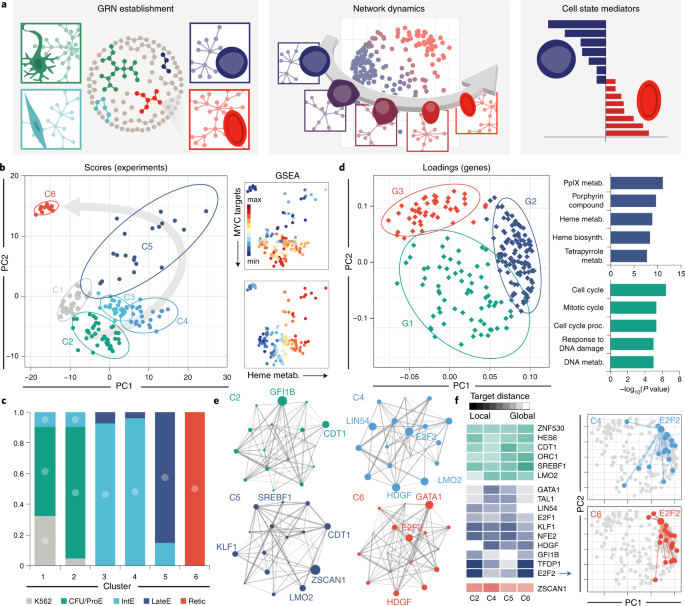
The fundamental roots in the CellNet algorithm evolved as we pursued a series of questions aimed at digging progressively deeper into the nuances of blood phenotypes. Starting broadly, we looked at differentiation from a binary perspective and asked: can we expand CellNet to learn more about hematopoiesis? We found that the network biology and machine learning analytics were able to distinguish hematopoietic progenitors from their red blood cell, or erythroid, derivatives. Although this is just one example among a plethora of successful CellNet applications, our analysis underscored the power and flexibility of the algorithm.
Next, narrowing in on the single lineage of erythropoiesis, we asked: is there a unique gene regulatory network signature corresponding to each intermediate cell type? This is where the biology forced us to depart from CellNet; we found that we could not simply reuse the same machine learning approaches to further parse out the stages of differentiation. Our attempts taught us that there were no mutually exclusive, isolated gene networks demarcating individual cell states. In evaluating new approaches, we turned to the systems biology expertise of the Lauffenburger lab at MIT. Inspired by recent innovations in single cell genomics, we used dimensionality reduction to view differentiation as a trajectory along a continuum. When combined with the additional layer of network biology information from CellNet, our new computational framework allowed us to analyze how networks were turned on and off and rewired during differentiation. This gave us a revealing snapshot of WHAT the network architecture looked like over time.
In the final stage of our pipeline, we asked WHY networks change between cell states. In other words, could we predict the signaling processes that govern differentiation? The key here turned out to be the establishment of a gene signature distinguishing two stages of differentiation – in this case, the maturation of erythroblasts to reticulocytes, a cell type that remains largely elusive in cell culture. We hypothesized that there were common master regulator(s) upstream of the disparate biological processes represented in the gene signature. We first started looking for master transcription factors, as they are commonly manipulated in cell engineering. However, the data, again, did not conform to our hypothesis. We were unable to find a “consensus” set of transcription factors controlling our differentiated gene signature, forcing us to look upstream at protein-protein signaling.
The protein-protein networks that we constructed from our differentiation signature pointed us toward several interesting signaling pathways, several of which had known roles in red blood cell development. However, one pathway kept reappearing in different analyses: ErbB signaling. This was particularly interesting because its involvement in blood seemed counterintuitive. In my colloquial view, EGFR/ErbB signaling was involved with cancer. However, when we started digging deeper into the biology, we discovered that the less studied family member, ErbB4, was involved in several developmental processes and, in our hands, required for robust blood generation. We then produced our own data by perturbing ErbB signaling and took that back to the model to explore the pathway further, ultimately linking ErbB4 to Wnt signaling. By identifying Wnt, a druggable target in the pathway, we were able to bring our hypotheses back into the stem cell culture dish and accomplish our original goal of improving the differentiation of red blood cells from pluripotent stem cells.
One major challenge of this project was striking a balance between the need to produce generalizable, unbiased computational models with the benefits of providing expert supervised interpretation. Broaching the challenge with an interdisciplinary team of biologists and engineers allowed me to see firsthand the power of moving freely across the computational-experimental space; taking the project all the way from model to hypothesis to validation in model organisms and back to molecular mechanisms in vitro reinforced the notion that computational modeling is not a unidirectional process.
Although I am excited to learn more about ErbB signaling in blood, this is just one very specific example; our pipeline is generally compatible with different data types, including RNA-seq, and applicable to other cell systems. We anticipate that our example will serve as a roadmap for future studies of dynamic biological processes, by highlighting critical elements to focus on (e.g., LASSO gene signatures) and how to connect them to pathways/processes (e.g., networks derived via PCSF and correlation), ultimately providing new hypotheses about the master regulators of cell fate and identifying cognate perturbations for cell engineering.
OUR PAPER: Kinney M.A., Vo L.T., Frame J.M., Barragan J., Conway A.J., Li S, Wong K., Collins J.J., Cahan P., North T.E., Lauffenburger D.A., Daley G.Q. “A systems biology pipeline identifies regulatory networks for stem cell engineering.” Nature Biotechnology. (2019). DOI: 10.1038/s41587-019-0159-2
Full text available at Nature Biotechnology: https://www.nature.com/articles/s41587-019-0159-2
--------
REFERENCES
[1] Cahan, Patrick, et al. “CellNet: Network Biology Applied to Stem Cell Engineering.” Cell, vol. 158, no. 4, Aug. 2014, pp. 903–15. doi:10.1016/j.cell.2014.07.020
[2] Morris, Samantha A., et al. “Dissecting Engineered Cell Types and Enhancing Cell Fate Conversion via CellNet.” Cell, vol. 158, no. 4, Aug. 2014, pp. 889–902. doi:10.1016/j.cell.2014.07.021
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in