Retinol-mediated suppression of tumor-promoting inflammation is disrupted in colon cancer-associated fibroblasts
Published in Cancer
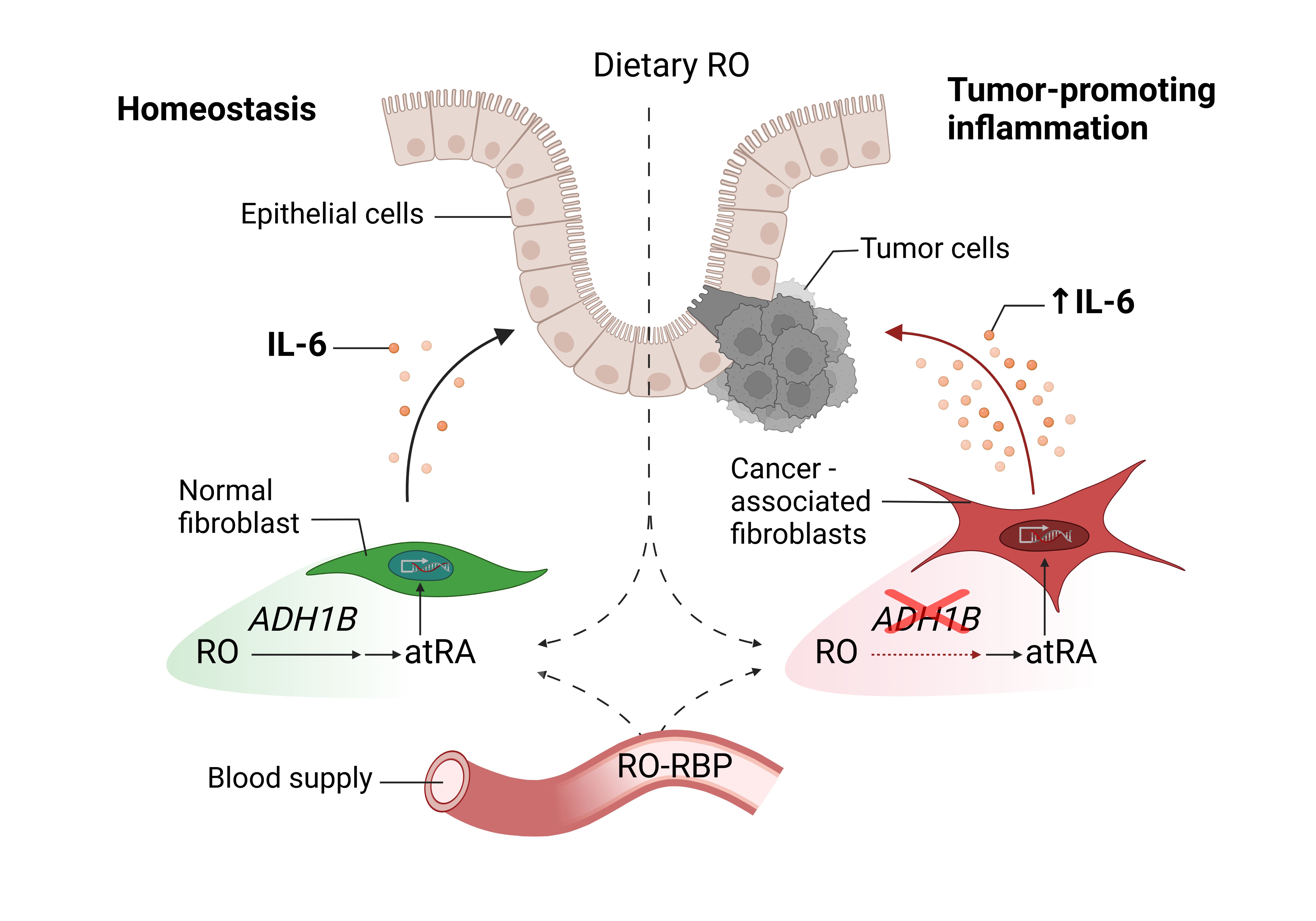

Colorectal cancer is one of the most common cancers worldwide1. However, the major mechanisms promoting tumor growth and cancer-associated inflammation are only partially determined, whereas knowledge of factors involved in cancer development is a critical issue for developing new treatments. The tumor microenvironment (TME) has emerged as an important target for cancer therapy because of its key role in tumor growth and immune escape through tumor-promoting inflammation2. Among the critical cells contributing to tumor inflammation, cancer-associated fibroblasts (CAFs) are very abundant of the TME and are major producers of IL-63,4. IL-6 is a cytokine expressed at high levels in the tumor microenvironment and is a major mediator of inflammation4,5. Increased IL-6 production by CAFs has been shown to promote tumor cell growth, cancer stem cells, and metastasis in colon cancer3,4,6. Targeting IL-6 is proposed as an attractive strategy in the treatment of GI cancers, but the mechanisms involved in the increase of this tumor-promoting cytokine are unknown. In this study, we intended to identify novel targets involved in the dysregulation of IL-6 expression by CAFs in colon cancer.
We first isolated colon cancer CAFs and normal fibroblast (N-MFs) from patient tissues7. Transcriptomic analysis of cultured CAFs demonstrated significant changes in the network of genes involved in the immune responses, angiogenesis, regulation of cytokine signaling network and metabolism when compared to cultured N-MFs. Among the genes dysregulated in CAFs, a striking decrease was observed in alcohol dehydrogenase 1B (ADH1B), a class I alcohol dehydrogenases involved in metabolism of alcohols, while upregulation of IL-6 was confirmed. However, the pathophysiological contribution of the decreased ADH1B expression to the increase in IL-6 and cancer progression remained unclear.
Similar observations were made in situ. We identified MFs by expression of α-SMA and we observed that ADH1B expression was mainly co-localized with N-MFs in normal tissues, while it was absent from the cancer stroma. To investigate the stage of neoplastic transformation where the loss of ADH1B gene expression occurs, we analyzed ADH1B mRNA expression in tubular adenomas, which represent an early neoplastic transformation stage, and at the colon cancer carcinoma (T1–T4) stage and compared these to the ADH1B level in normal colonic tissue. We also determined the expression of this gene in benign hyperplastic polyps. A significant decrease in ADH1B expression was observed in approximately 50% of tubular adenoma studied and in all T1-4 colon carcinomas, but not in hyperplastic polyps, suggesting that ADH1B is drastically decreased during the neoplastic to carcinogenic progression.
We and others have previously demonstrated that an increase in basal and inducible IL6 production by CAFs is among key factors promoting tumor progression in colon cancer3,4, but the causes of this increase remain unknown. To evaluate whether ADH1B is involved in the regulation of IL-6 production, the expression of Adh1b was knocked down in N-MFs using specific siRNA, then cells were treated with pro-inflammatory lipopolysaccharide (LPS). The silencing of Adh1b in N-MFs resulted in a dramatic increase in LPS-induced IL-6 expression and secretion.
ADH1B is a key enzyme inf the metabolism of retinoids. Retinoids are biologically active derivatives of vitamin A (retinol), a fat-soluble vitamin absorbed from the intestinal tract and mainly stored in the liver as retinyl esters 8. The intracellular processing of retinol involves several enzymatic steps to exert biological activities. Retinol (RO) is reversibly oxidized into retinaldehyde (RAL) by alcohol dehydrogenases or retinol dehydrogenases. Then RAL is irreversibly metabolized by aldehyde dehydrogenases, generating all-trans retinoic acid (atRA), the most active retinoid metabolite. atRA regulates various biological processes such as cell differentiation, proliferation and apoptosis. atRA interaction with nuclear retinoic acid receptors (RARs) and retinoic X receptors (RXRs) may activate or repress the transcription of various target genes through binding to DNA-retinoic acid response elements (RAREs)9. Retinoids are known to inhibit tumor-promoting IL-6 production 10, but the regulating mechanisms have not been fully explored. Moreover, although a therapeutic anti-tumor effect of atRA has been shown in several cancers, their use for treatment of solid tumors has yielded controversial results11. We hypothesized that ADH1B may convert retinol to retinaldehydes, which are metabolized to atRA, which in turn could negatively regulate IL-6 production. Therefore, we next evaluated the consequences of the low levels of ADH1B in CAFs on IL-6 production after retinoid treatment, and compared this to IL-6 production in N-MFs that exhibit high levels of ADH1B. In N-MFs, LPS-induced IL-6 production was inhibited by either RO or its byproduct atRA. However, although atRA was effective in CAFs, RO was not able to reduce LPS-induced IL-6 production, confirming the inability of CAFs to utilize RO to suppress IL-6 production. While other mechanisms may be responsible for the IL-6 increase within the CRC tumor stroma, these results highlight the crucial role of a stromal vitamin A pathway in its regulation.
To conclude, this work demonstrated that an abrogation of ADH1B expression in MF during the adenoma-carcinoma sequence promotes IL-6-driven tumor-promoting inflammation in CRC that involved the disruption of the vitamin A-mediated regulation of IL-6. Taken together, our data identified ADH1B as a novel potential mesenchymal suppressor of tumor-promoting IL-6 overexpression.
References
- Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet Lond. Engl. 394, 1467–1480 (2019).
- Schmitt, M. & Greten, F. R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 21, 653–667 (2021).
- Nagasaki, T. et al. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br. J. Cancer 110, 469–478 (2014).
- Huynh, P. T. et al. CD90 + stromal cells are the major source of IL-6, which supports cancer stem-like cells and inflammation in colorectal cancer: Fibroblast derived IL-6 supports CRC. Int. J. Cancer 138, 1971–1981 (2016).
- Johnson, D. E., O’Keefe, R. A. & Grandis, J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018).
- Zhu, L. et al. Bone marrow-derived myofibroblasts promote colon tumorigenesis through the IL-6/JAK2/STAT3 pathway. Cancer Lett. 343, 80–89 (2014).
- Johnson, P. et al. Isolation of CD 90+ Fibroblast/Myofibroblasts from Human Frozen Gastrointestinal Specimens. J. Vis. Exp. 53691 (2016) doi:10.3791/53691.
- Blomhoff, R. & Blomhoff, H. K. Overview of retinoid metabolism and function. J. Neurobiol. 66, 606–630 (2006).
- Tang, X.-H. & Gudas, L. J. Retinoids, Retinoic Acid Receptors, and Cancer. Annu. Rev. Pathol. Mech. Dis. 6, 345–364 (2011).
- Kirchmeyer, M. et al. All-trans retinoic acid suppresses interleukin-6 expression in interleukin-1-stimulated synovial fibroblasts by inhibition of ERK1/2 pathway independently of RAR activation. Arthritis Res. Ther. 10, R141 (2008).
- Costantini, L., Molinari, R., Farinon, B. & Merendino, N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. J. Clin. Med. 9, 360 (2020).
Follow the Topic
-
British Journal of Cancer
This journal is devoted to publishing cutting edge discovery, translational and clinical cancer research across the broad spectrum of oncology.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in