Revealing how dysregulation of GPCR signaling can lead to human disease
Published in Healthcare & Nursing
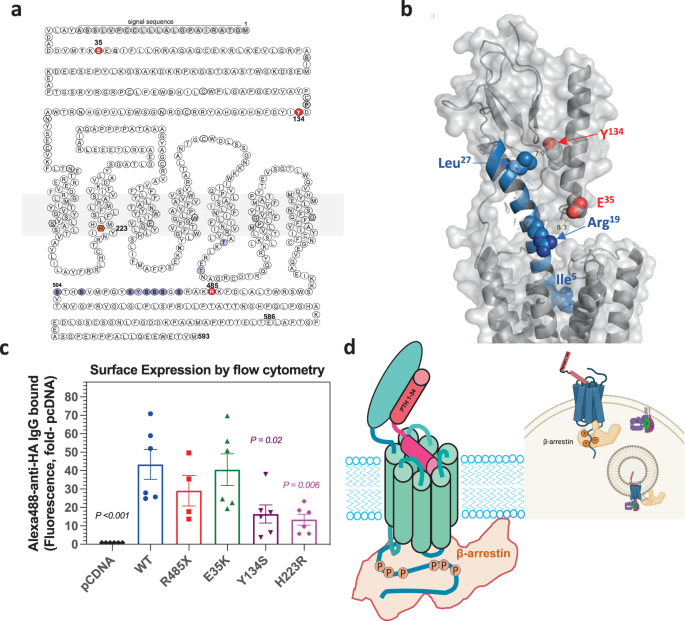
G protein coupled receptors (GPCRs) are cell membrane spanning proteins that play critical roles in biology by relaying information carried in external stimuli, such as a hormone, a neurotransmitter, or even a particle or waveform of light, from the outside of a cell to the cell interior where a “second messenger” mediator is activated, and an intended functional response occurs. In general, for an optimal adaptive physiological response to occur, the process of transmembrane signal transduction must be precisely regulated, in terms of limiting the amplitude and duration of signal transmission. Such GPCR signal regulation results in a desensitization of the system to the initial stimuli, and occurs via multiple subcellular events, including the recruitment of βarrestin proteins to cytoplasmic surfaces of the receptor and the internalization of the GPCR signaling complex to endocytic vessels-- processes that result in signal termination.
The ~ 800 GPRCs in humans are divided based on amino acid sequence homology into six main classes--A, B1, B2, C, F and others. The parathyroid hormone receptor type 1, PTH1R, is a class B1 GPCR that regulates two distinct physiological processes -- bone development and calcium homeostasis, which it does by binding two distinct peptide hormone ligands, PTH and PTHrP, respectively.
Eiken syndrome is a rare disease of skeletal development, characterized by delayed bone mineralization. This clinical phenotype, readily apparent on x rays of the hands of young patients, suggests an exaggerated PTH1R signaling response, particularly to PTHrP, because the receptor and PTHrP normally act in the growth plates of developing bones to control the mineralization process and hence to prevent premature ossification. Previous genetic analyses of three patient families with Eiken syndrome revealed three different homozygous missense mutations in the PTH1R of affected individuals. How these mutations impact PTHR1 function, however, was unexplored.
In our new study, we characterized the three Eiken PTH1R mutants using transfected HEK293 cells and a variety of experimental assay systems to investigate receptor function, including signaling, internalization to endosomes and recruitment of βarrestin proteins. Our results reveal that one of the mutations, R485X, which truncates the PTH1R cytoplasmic tail and hence removes key serine/threonine phosphorylation sites thought to be needed for efficient βarrestin recruitment, indeed impairs βarrestin recruitment and hence results in elevated ligand-independent levels of basal signaling via the cAMP second messenger pathway.
The two other mutations, E35K and Y134S, alter sites in the receptor’s extracellular domain, the ECD, which plays a key role in binding the extracellular PTH and PTHrP ligands. Analysis of the two mutant receptors revealed little or no effect on basal cAMP signaling, as compared to WT PTH1R; however, the complexes that the mutant receptors formed with PTHrP were less stably than those formed by PTH1R WT, and as result the two mutants exhibited defective capacities to recruit βarrestin to endosomes and to desensitize their response to PTHrP ligand exposure. Interestingly, changing just one key amino acid in PTHrP (His>Ile at position 5), rescued the beta arrestin recruitment for E35K and Y134S but not for R485X, highlighting the requirement of precise interactions between PTH1R and PTH/PTHrP for normal βarrestin recruitment.
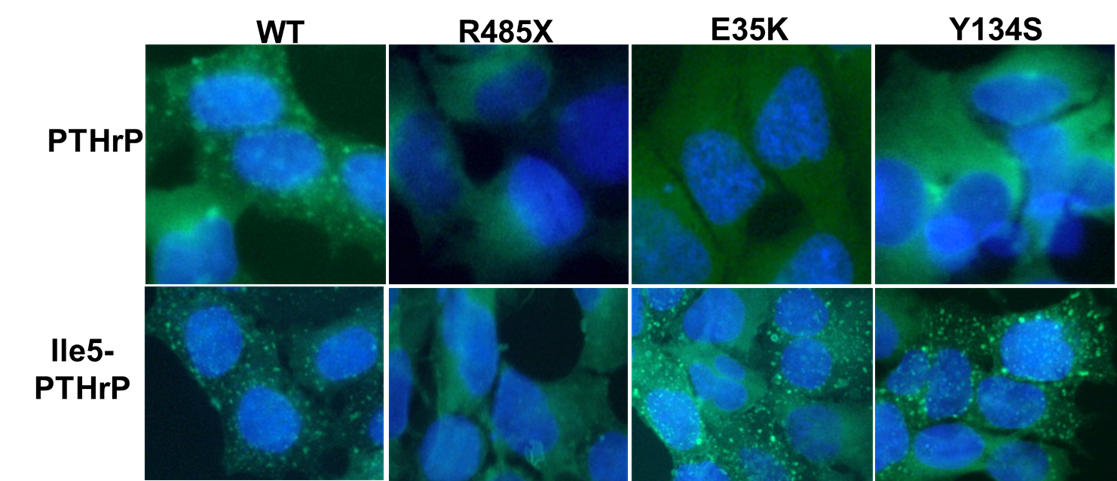
Because PTHrP is produced locally in the growth plates of developing bones and acts as a paracrine factor to regulate the differentiation of adjacent primordial growth plate chondrocytes, a defect in the capacity of a mutant PTH1R in those target cells to desensitize the response to PTHrP could result in an exaggerated signaling response and hence a delay in the bone mineralization process, as observed in the Eiken Syndrome patients with the E35K and Y13S PTH1R mutations. A caveat to our studies is the use of peptide fragments and not full length native PTHrP.
The findings overall implicate an important role for GPCR desensitization mechanisms, and particularly for the capacity of a class B1 GPCR to form a complex with its peptide hormone ligand that is stable enough to enable efficient recruitment of βarrestin proteins, in normal biology, and they highlight how even slight perturbation of these mechanisms can result in human disease.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
From RNA Detection to Molecular Mechanisms
Publishing Model: Open Access
Deadline: May 05, 2026
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Jun 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in