SARS-CoV-2 infection of human lung epithelial cells induces TMPRSS-mediated acute fibrin deposition
Published in Microbiology
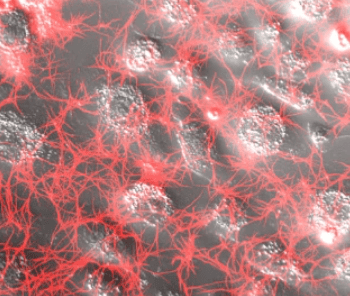

Background
In the early days of the COVID-19 pandemic, little was known about the pathogenesis of SARS-CoV-2, except that the mortality associated with the infection was much higher than seasonal flu and many hospitalized patients required mechanical ventilation. Early autopsy showed extensive alveolar damage, fibrin aggregation, and micro-thrombotic structures in infected lungs. These autopsy results showed a possible dysregulation in the coagulation pathway and many clinicians and hospitals experimented with various anti-coagulation treatments. The publications from these treatments, however, showed mixed clinical efficacy. Faced with mounting mortality associated with the pandemic and a desire to control the public health crisis, a coordinated large-scale multi-national clinical trial took place to evaluate clinical efficacy of using systemic injection of low molecular weight heparin as an anti-coagulant to reduce mortality associated with severe COVID. In many ways, this trial was a beacon in the thick of the pandemic shrouded by images of crowded mass graves and long lines of cremation of the deceased. It was therefore, rather disappointing that the clinical trial ended on futility criteria (N Engl J Med 385:777-89, 2021), meaning the systemic injection of the anticoagulation drug did not mitigate the severe COVID associated mortality. The failure in anticoagulation therapy was in stark contrast to the success in SARS-CoV-2 vaccine and anti-viral drug developments.
Among the three pillars of COVID public health measures— vaccine, antiviral, and therapeutic treatment—therapeutics lagged behind. Many investigators in the US and the world had already engaged in intense research in both vaccine and antiviral development. In contrast, therapeutic treatments were relatively less developed and the failure of the low molecular weight heparin trial, published in the New England Journal of Medicine, discouraged many commercial drug companies from developing a similar line of treatment products. There was a clear need to understand the mechanism of SARS-CoV-2-induced lung pathogenesis and we thought that understanding the mechanism was the first step for developing a therapeutic approach and mitigating severe COVID. Our lab’s research background was in HIV-1 pathogenesis, and we felt in the early pandemic (year 2020) that our knowledge of HIV-1 was a good fit for studying SARS-CoV-2 pathogenesis. Motivated by the severity of the pandemic, we began to investigate the mechanism of viral infection-induced fibrin depositions in the lung.
Early on in the pandemic, it was widely accepted that SARS-CoV-2-associated lung pathogenesis was largely due to viral-induced lung inflammation, as the presence of pneumonia in infected lungs was ubiquitous in both human and animal models. The subsequent inflammatory responses by immune systems, such as cytokine infiltrations, would result in damage to the infected lungs, the essence of immunopathology. Several animal models since developed for COVID research, including mice, Syrian hamsters, and monkeys, however, seem to tell a somewhat different story. While all animals are susceptible to SARS-CoV-2 infections and are great models for vaccine and antiviral development, most of infected animals displayed only mild symptoms and recover in two weeks. Animals infected with a normal dose rarely die. Pathology findings also presented significant distinction between human diseases and animal models. While both human and animals developed viral pneumonia in response to SARS-CoV-2 infections, diffuse alveolar damage (DAD) as characterized by frequent observation of hyaline membranes and fibrin deposition are common in infected human lungs but rare in infected animals.
Developing a Model System
The first issue was to establish a suitable model system. Even though the development of lung pathology is an in vivo phenomenon, the existing animal models appeared inadequate to address the development of DAD. We then explored the use of primary human lung epithelial cells, both normal human bronchoalveolar epithelial (NHBE) and human small airway epithelial (HSAE) cells as infection models. In addition to the cell model, we needed to develop a viral infection system and fibrin deposition assay to address the link between the viral infection and fibrin deposition. Part of the challenge about SARS-CoV-2 is that it was an emerging pathogen with very little background information, and we had to integrate new ideas and information as they were published in real time. Fortunately, there were several versions of pseudovirus constructs available specifically for SARS-CoV-2 and one of the widely used pseudo-SARS-CoV-2 construct combined SARS-CoV-2 spike gene with an HIV-1 core plasmid to generate a hybrid, replication incompetent pseudo-SARS-CoV-2 virus. The advantage of this construct is the spike gene can be easily replaced to address various evolving strains of SARS-CoV-2. For the fibrin deposition, we adopted an in vitro turbidity-based fibrin clotting assay to measure the cleavage of fibrinogen and subsequent polymerization of fibrin. Once we validated the infectivity of our pseudovirus, we combined the pseudo-SARS-CoV-2 infection of primary human epithelial lung cells with the fibrin clotting assay.

NHBE cells trigger fibrin clot formation.
To our surprise, we observed that infected cells triggered fibrin clotting and uninfected cells did not (Figure 1). The surprise is that this SARS-CoV-2 infection-induced fibrin aggregation occurred in the absence of traditional components of the classical coagulation pathway, suggesting involvement of cellular proteases.
In the classical coagulation pathway, fibrinogen is cleaved into fibrin by activated thrombin. Although we did not add any thrombin or other proteases to our in vitro infection-induced fibrin clotting, we found that the infection-induced fibrin formation was inhibited by thrombin inhibitor, hirudin, suggesting the presence of thrombin. Further mass spectrometry-based protein identification on infected cell supernatants revealed the presence of bovine thrombin in the cell culture supernatant, likely from bovine serum-based cell culture media formulation.

caused no clot formation, but clotting was rescued
with the addition of human prothrombin.
Further, depletion of bovine prothrombin with an anti-thrombin antibody abrogated the SARS-CoV-2 infection-induced fibrin clotting, and the addition of human prothrombin to the bovine prothrombin depleted media rescued the fibrin clotting (Figure 2). Thrombin circulates as inactive prothrombin in plasma and must be activated by the upstream coagulation factor Xa as part of the classical coagulation pathway.
Investigating the Mechanism

is sufficient to induce fibrin clotting.
We wondered how prothrombin was activated during SARS-CoV-2 infection of lung epithelial cells. Based on our observation that supernatant from infected lung cells was sufficient to induce fibrin clotting (Figure 3), we then tested the ability of SARS-CoV-2 infected supernatant to cleave the prothrombin peptide, a necessary step in prothrombin activation. Indeed, the viral infected supernatant showed higher enzymatic cleavage of the prothrombin peptide than the uninfected supernatant.
These observations led us to further investigate which cellular genes contribute to non-classical coagulation induced by the viral infection of lung epithelial cells. We noticed that not all SARS-CoV-2 infected cells support fibrin clotting. While both infected NHBE cells and infected HSAEC supported fibrin clotting, infected Vero E6 and ACE2-293T cells did not form fibrin clots.

The lack of fibrin clot formation is not due to their infection level as both Vero E6 and ACE2-293T cells are very susceptible to SARS-CoV-2 infections. Interestingly, the fibrin clotting ability of infected cells appear to correlate with their expression of members of the type 2 transmembrane serine proteases (TTSP), also known as TMPRSS genes. Both the clotting cells, NHBE and HSAEC, express ST14 and TMPRSS11D genes whereas the non-clotting Vero E6 and ACE2-293T cells lack TTSP expressions. RNA seq analysis revealed a mild upregulation in ST14 and TMPRSS11D expressions. We then hypothesized that viral infections activate the serine proteases of TTSP, resulting in the bypass of classical coagulation pathway and the cleavage of prothrombin by activated TTSP, leading to fibrin deposition. The protease domains of ST14 and TMPRSS11D are referred to as matriptase and human airway trypsin-like protease (HAT), respectively.

peptide in vitro.
Both matriptase and HAT cleaved the prothrombin peptide and contribute to fibrin clotting in vitro (Figure 5). Although tissue factor of the classical coagulation pathway was reported upregulated in SARS-CoV-2 infected cells, we did not observe its upregulation in infected NHBE nor infected HSAEC cells. Nevertheless, we knocked out the tissue factor in both NHBE and HSAEC cells using CRISPR/Cas9.

the classical coagulation pathway, in primary human
lung cells.
Despite a near complete knockout of the tissue factor (Figure 6), the infected cells triggered fibrin clot formation, demonstrating that the infection-induced fibrin clotting is independent of the classical coagulation initiator, tissue factor.
To address if the expression of matriptase or HAT on cells was sufficient to trigger infection-induced fibrin clotting, we transfected non-clotting ACE2-293T cells with plasmids encoding full-length genes, then infected the transfected cells with SARS-CoV-2 pseudovirus for fibrin clotting assays. The results showed that infected cells expressing matriptase or HAT gained the ability to generate fibrin clots. Together, these results show that SARS-CoV-2 infection of lung epithelial cells induced shedding of TTSP proteins, such as matriptase and HAT, that are capable of activating prothrombin for fibrin clot formations.
Ex vivo Studies
At this point, we had a good cell-based in vitro model system to show SARS-CoV-2 infection induced fibrin clot formation. All our observations were also reproduced with replication competent field strains of SARS-CoV-2 viruses, including Washington strain, alpha, beta, delta, and omicron strains. However, the big question was if the infection-induced fibrin deposition occurred in infected human lungs in vivo. Since the existing animal models did not show significant clinical lung pathology, especially the appearance of DAD, we performed an ex vivo experiment to evaluate the likelihood of fibrin deposition induced by SARS-CoV-2 infection in human lungs. As prothrombin and fibrinogen are plasma proteins, it is likely that their presence in infected lungs were the result of inflammatory infiltration, which is also responsible for the presence of neutrophils, T cells, macrophages and other immune cells in infected lungs. Since the trigger of the fibrin deposition depends on prothrombin activation by cell surface expressed serine proteases, we then asked 1) if the concentrations of prothrombin and fibrinogen are elevated in SARS-CoV-2 infected lung fluid and 2) if infected lung fluid supports fibrin clotting in the presence of the viral infection. To address these questions, we performed mass spectrometry-based proteomic analyses on bronchoalveolar lavage fluid (BALF) samples from both healthy and COVID patients. We found that the majority of COVID research groups focused on plasma samples and only few investigators made attempt to collect BALF samples by an invasive bronchoscope procedure. Fortunately, Dr. Twigg III, professor of medicine from Indiana University medical school who runs a pulmonary clinical laboratory had collected BALF samples from acute and recovered COVID individuals and was willing to collaborate on the topic.Our proteomic analyses together with ELISA experiments showed an increased infiltration of plasma components such as fibrinogen and prothrombin in COVID lungs, and that the concentrations of these components, especially fibrinogen and prothrombin, were upregulated 50-100 fold in acute COVID samples compared to those of healthy BALF.

Importantly, infected but not uninfected lung epithelial cells produced fibrin clots in the presence of BALF from acute COVID patients, suggesting the levels of prothrombin and fibrinogen in acute COVID BALF are sufficient to support SARS-CoV-2 infection induced fibrin clotting (Figure 8).

No fibrin clotting was observed from infected NHBE cells in the presence of BALF from healthy individuals. Interestingly, not all acute COVID BALF supported fibrin clotting. While three out of four acute COVID BALF supported fibrin clotting, one sample did not despite the presence of elevated prothrombin and fibrinogen, consistent with not all severe COVID patients developed lung fibrosis and suggesting other factors, such as plasminogen, antithrombin III, and other fibrinolytic factors may also contribute to fibrin deposition in infected lungs.
Conclusion
In summary, we showed that SARS-CoV-2 infection of human lung epithelial cells induced fibrin deposition in the presence of fibrinogen and prothrombin. It is worthwhile to note the differences between this viral-induced fibrin deposition and that from classical coagulation. In classical extrinsic coagulation pathway, a coagulation is initiated by tissue factor and depends on a sequential activation of several plasma serine proteases. This results in the activation of factor X to produce factor Xa, which cleaves prothrombin peptide, leading to thrombin activation. In SARS-CoV-2-induced coagulation, the viral infection activates transmembrane serine proteases on the infected cells, bypassing the cascade of plasma serine proteases. This leads to cleavage of prothrombin and subsequent fibrin deposition. Importantly, classical coagulation occurs in plasma and the viral-induced coagulation depends only on plasma prothrombin and fibrinogen but occurs in the alveolar airway outside of circulating plasma. The presence of elevated prothrombin and fibrinogen levels in the infected alveolar space is presumably due to inflammatory infiltration associated with the viral infection (Figure 9).

In principle, the cascade of plasma serine proteases in classical coagulation pathway should also be present in infected alveolar airways. Their concentrations are much lower compared to prothrombin and fibrinogen, likely insufficient to induce fibrin clotting as evidenced from the lack of fibrin clotting in acute COVID BALF in the absence of the viral infection. It is conceivable that the presence of the viral-induced fibrin clotting either initiates or exacerbates fibrin deposition in infected lung. Our findings provide an explanation for the failure of the multi-national heparin trial in reducing severe COVID mortality. As the heparin clinical trial and many early hospital treatments focused on inhibiting plasma coagulation, the treatments may be less effective to SARS-CoV-2 infection-induced alveolar coagulation outside of blood circulation.
While our work specifically addressed fibrin deposition induced by SARS-CoV-2 infections, it begs the broader question of if the findings also apply to other viral infections, such as seasonal flu, MERS, and other respiratory infections. Fundamentally, we would like to know if this is generally applicable to viral infection-associated acute respiratory distress syndrome (ARDS) as similar alveolar damage has been observed in other severe respiratory infections. We speculate the mechanism for viral infection-induced fibrin deposition is more general. This is supported by our observation that infection of NHBE cells with a VSV-typed pseudovirus also induced fibrin clotting. As mentioned earlier, the existing animal models, including mouse, Syrian hamster, and monkey, do not reflect human severe COVID-associated lung pathology. As a result, there is no suitable in vivo model for therapeutic treatment. We hope that our work will open up new avenues for animal models that ultimately applicable to therapeutic research.
Taken together, the results of this study reveal a novel non-classical coagulation pathway of acute fibrin deposition induced by SARS-CoV-2 infection. Because the prothrombin activation depends on cellular proteases, targeting plasma coagulation with systemic injection of heparin-like anticoagulants may not be effective as the viral-induced fibrin clotting occurs in alveolar airways outside of circulating plasma. We suggest a better therapeutic approach is to target SARS-CoV-2 infected lungs directly with thrombin inhibitors.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in