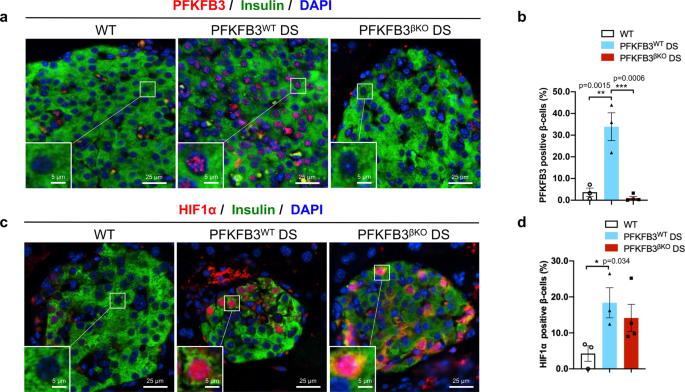
Aging post-mitotic cells and tissues are subjected to a variety of stressors, of which misfolded proteins and endoplasmic reticulum stress play a major part. Our group is interested in the effect of such stresses on a complex glandular organ like the pancreas1, 2, and has generated compelling evidence supporting the premise that cell fitness competition is an essential regulator of the health and function of the adult endocrine pancreas 3. Cell fitness competition is emerging as a critical mechanism of post-mitotic tissue homeostasis; it is a process that embodies the constant monitoring and culling of aged, injured, or dysfunctional cells in adult tissue so that healthier and functional cells can thrive4. In a multi-cellular society, fitness is the metric of wealth, enabling “fit” cells to monopolize space and resources at the expense of their “less fit” neighbors. The process relies on detecting the differential expression of quality control proteins across several essential pathways—from energy production and metabolism to replication—in what has been termed the “fitness fingerprint.” In a healthy system, cells expressing the “loser” fitness fingerprint are quietly removed and replaced by healthy (“winner”) cells through replication and regeneration, so that the overall tissue size doesn’t change but cell fitness is maintained. As such, the presence of β-cell competition may also explain a negligible rate of replication in human adult β-cells in endocrine pancreas. Unlike some organs, such as the skin, where cells undergo high turnover constantly (high replication and high rates of cell death), in the healthy pancreas, the low rate of β-cell replication is reflective of the low rate of injury/death. Besides, given that replication during cell fitness competition would occur in a compensatory manner relative to the rate of cells undergoing cell death, the overall result will have no effect on the β-cell mass. This is precisely what we observe in type-2 diabetes (T2D), whereas the overall β-cell mass doesn’t change dramatically, but the pancreatic function becomes compromised as unpurged, injured β-cells accumulate. Interestingly, proteotoxicity (such as caused by toxic oligomers of islet amyloid pancreatic polypeptide, IAPP) that accumulates with aging was previously affiliated with the “loser cell” status5. We also previously demonstrated that 30–60% of all β-cells from patients with diabetes have abnormalities that mirror the fitness fingerprints of loser (damaged) cells, with a predominant feature being proteotoxicity and metabolic remodeling by the HIF1α-PFKFB3 injury/repair pathway3, 6, 7. However, instead of these cells being targeted and removed as usual, this extrinsic control of tissue function and regeneration apparently falls short in the context of chronic stress, leading to the persistent accumulation of glucose-intolerant β-cells. Our model postulates that cell fitness comparison is, essentially, a gatekeeper to pancreatic tissue homeostasis and regeneration, by which: (1) misfolded protein stress favors a population of predominantly loser cells; (2) chronic stress from environmental factors, i.e., obesity, accumulates and eventually impedes or mitigates the culling of loser cells8; and (3) the coupling of step 1 and step 2 leads to an imbalanced pancreatic islet system, flooded with loser cells that are no longer responsive to metabolic indicators and that threaten pancreatic islet function and onset of disease.
It is not surprising that metabolism exerts a role in determining the survival advantage of the loser cells, given that energy is equivalent to life and growth. The combined effects of HIF1α and PFKFB3 govern, essentially, the “monetary” currency of the cell, as they mediate the oxygen-independent (e.g., mitochondria-independent) capacity to generate energy. These insights motivated us to investigate the links between β-cell competition and disease (i.e., diabetes), using a mouse model that recapitulates human disease following the β-cell-specific transgenic expression of hIAPP (proteotoxicity) and administration of a high-fat diet (which causes metabolic stress akin to obesity)3. The combination of proteotoxicity and high-fat diet simulates diabetogenic stress (DS), similar to what is seen in humans that develop diabetes. Using this model, we demonstrated that the β-cell-specific disruption of the Pfkfb3 gene led to the restoration of islet mass and function with an extent dependent on the relative level of DS and HIF1α. For instance, after we triggered chronically low levels of DS, PFKFB3-knockout animals experienced a removal or culling of the β-cells with loser fitness fingerprint, followed by a wave of healthy β-cell replication (comparable to WT mice), with subsequent restoration of β-cell mass and function. The regenerating wave of “healthy” β-cells did not appear to restore function under high DS3. These differential outcomes appear to hinge on the independent expression of HIF1α in our model system, underscoring the notion that there is a functional dichotomy that exists between HIF1α and its target, PFKFB3, which serve in tandem to govern the mechanisms that are responsible for re-establishing a fully functional complex tissue after part of the β-cell mass is eliminated due to cell competition.
The fact that the upregulation of HIF1α and PFKFB3 coincide with a loss of β-cell function in prediabetes further implicates the involvement of β-cell fitness competition mechanisms as early as the β-cell decompensation stage and support the premise that HIF1α and/or PFKFB3 inhibition would be a viable therapeutic strategy to prevent the onset of diabetes or potentially even reverse its progression in established disease. Aside from their potential therapeutic implications for T2D, these findings are significant, as they: (1) point to the existence of similar or different molecular matrices of cell fitness competition in other low-turnover tissues like pancreas; (2) implicate inhibition of cell competition as contributor to disease; and (3) could potentially be used to stimulate regeneration in adult, post-mitotic tissues.

References
- Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC. Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes. 2004;53(6):1509-16. doi: 10.2337/diabetes.53.6.1509. PubMed PMID: 15161755.
- Huang CJ, Haataja L, Gurlo T, Butler AE, Wu X, Soeller WC, Butler PC. Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab. 2007;293(6):E1656-62. Epub 20071002. doi: 10.1152/ajpendo.00318.2007. PubMed PMID: 17911343.
- Min J, Ma F, Seyran B, Pellegrini M, Greeff O, Moncada S, Tudzarova S. β-cell-specific deletion of PFKFB3 restores cell fitness competition and physiological replication under diabetogenic stress. Communications Biology. 2022;5(1):248. doi: 10.1038/s42003-022-03209-y.
- Merino MM, Levayer R, Moreno E. Survival of the Fittest: Essential Roles of Cell Competition in Development, Aging, and Cancer. Trends Cell Biol. 2016;26(10):776-88. Epub 20160616. doi: 10.1016/j.tcb.2016.05.009. PubMed PMID: 27319281.
- Baumgartner ME, Dinan MP, Langton PF, Kucinski I, Piddini E. Proteotoxic stress is a driver of the loser status and cell competition. Nat Cell Biol. 2021;23(2):136-46. Epub 20210125. doi: 10.1038/s41556-020-00627-0. PubMed PMID: 33495633; PMCID: PMC7116823.
- Montemurro C, Nomoto H, Pei L, Parekh VS, Vongbunyong KE, Vadrevu S, Gurlo T, Butler AE, Subramaniam R, Ritou E, Shirihai OS, Satin LS, Butler PC, Tudzarova S. IAPP toxicity activates HIF1alpha/PFKFB3 signaling delaying beta-cell loss at the expense of beta-cell function. Nat Commun. 2019;10(1):2679. Epub 20190618. doi: 10.1038/s41467-019-10444-1. PubMed PMID: 31213603; PMCID: PMC6581914.
- Nomoto H, Pei L, Montemurro C, Rosenberger M, Furterer A, Coppola G, Nadel B, Pellegrini M, Gurlo T, Butler PC, Tudzarova S. Activation of the HIF1alpha/PFKFB3 stress response pathway in beta cells in type 1 diabetes. Diabetologia. 2020;63(1):149-61. Epub 20191113. doi: 10.1007/s00125-019-05030-5. PubMed PMID: 31720731; PMCID: PMC6945783.
- Sasaki A, Nagatake T, Egami R, Gu G, Takigawa I, Ikeda W, Nakatani T, Kunisawa J, Fujita Y. Obesity Suppresses Cell-Competition-Mediated Apical Elimination of RasV12-Transformed Cells from Epithelial Tissues. Cell Rep. 2018;23(4):974-82. doi: 10.1016/j.celrep.2018.03.104. PubMed PMID: 29694905; PMCID: PMC6314181.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in