Tumour in normal contamination assessment from whole genome sequencing to ensure high standards of genomic testing and insights into personalised treatments for patients with haematological cancers
Published in Cancer, Protocols & Methods, and Computational Sciences
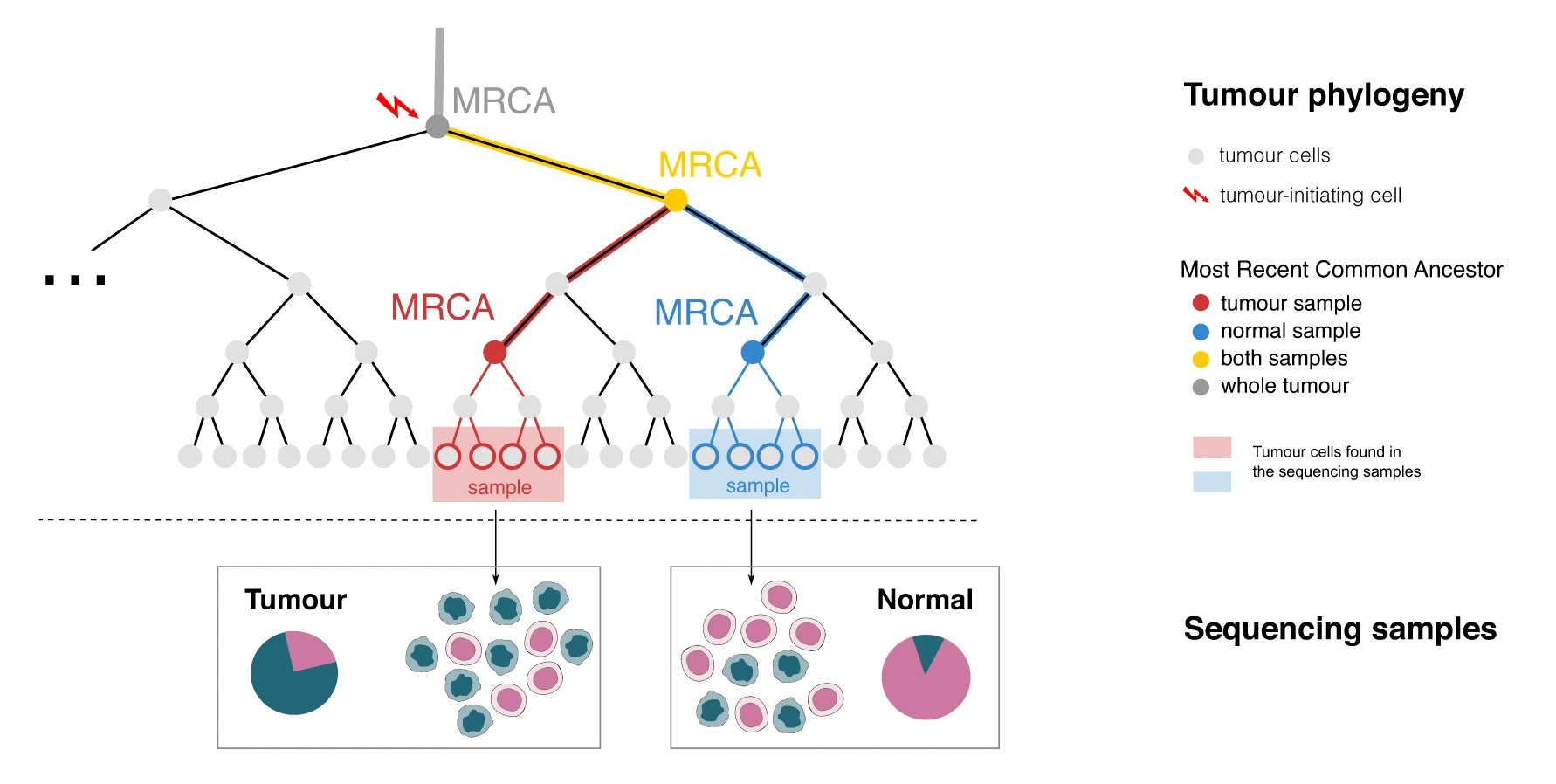
Genomic England provides whole genome sequencing (WGS) as a clinical test for NHS patients with different cancer types, including acute leukaemia. WGS plays a vital role in cancer care by providing a comprehensive genetic profile of the disease that can enable personalised treatment. It marks a significant shift towards effective precision cancer care. WGS is carried out as part of tumour molecular profiling, which requires samples from both tumour tissue and non-cancerous ‘normal’ tissue (usually blood). Typical tumour-normal analysis uses the normal sample to identify a patient’s germline variants, which are then subtracted from the variants identified in the tumour. This allows us to define the tumour-specific somatic variants.
The problem of contamination
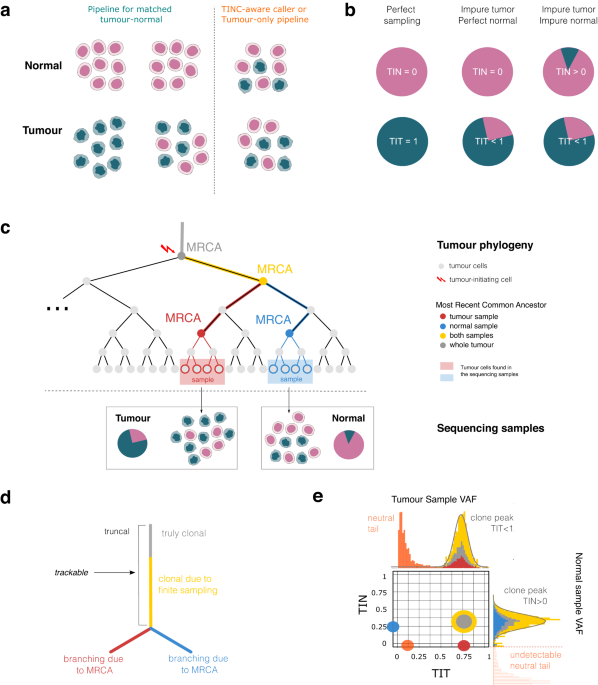
Bioinformatics pipelines designed for tumour-normal analysis are well-equipped to deal with tumour samples that have been contaminated with normal cells. But the opposite scenario, where normal tissue samples are contaminated by tumour cells, known as tumour in normal (TIN) contamination, can be problematic. TIN causes standard pipelines to unexpectedly subtract somatic variants, reducing variant detection sensitivity and compromising later analysis. This ultimately impacts the precision of tumour profiling in poor quality normal samples. TIN is particularly relevant in haematological cancers due to the natural spread of tumour cells within the bloodstream.
Making the most of a global research community and vast resource
Back in 2020, we found ourselves discussing TIN in a London pub and realised that we could estimate TIN contamination levels by tracking clonal somatic variants in the normal sample. Our recently published MOBSTER machine learning model for sub-clonal deconvolution from WGS (Nature Genetics 52, 2020) seemed the right tool to identify clonal somatic variants based on their variant allele frequency.
Following these conversations, we made the most of our WGS data for haematological tumours that was generated as a part of 100,000 Genomes Project, to develop a new computational open-source tool called TINC (https://caravagnalab.github.io/TINC/). TINC uses the principles of tumour evolution to estimate the level of TIN contamination from WGS assays, and generates a score for the percentage of tumour cells in the tumour and the normal sample. This score is simple to interpret, and can be computed automatically by integrating different data types. It allows an informed decision-making process regarding variant interpretation and reporting, or alternative variant calling procedures.
We were then able to do extensive testing and validate TINC using real-world patient data from the 100,000 Genomes Project stored in the National Genomic Research library. This data can be accessed by any approved researchers in our global research community, known as the Genomics England Research Network (see these instructions on how to join).
Working with our long-term collaborator Dr Jack Bartram, a leading consultant paediatric haematologist at Great Ormond Street Hospital, we were also able to validate TINC against standard disease-monitoring technologies for minimal residual disease testing. Now, our TINC tool is implemented in a clinically accredited bioinformatics pipeline for WGS tumour analysis at Genomics England, helping to identify contaminated normal samples. When TIN is detected, the pipeline which is optimised to process haematological tumours, uses alternative variant-detection strategies to ensure highly sensitive performance.
The wider impact of our work
By combining expertise in bioinformatics and machine learning from scientific experts at Genomics England and the Cancer Data Science Laboratory of the University of Trieste, with testing in clinical settings we’ve been able to create a tool with real-world impact for clinicians using WGS data to diagnose and treat patients with cancer.
Our TINC tool has now been implemented in a clinically accredited bioinformatics pipeline used by Genomics England for WGS tumour analysis to support the NHS, helping to identify contaminated normal samples and ensure high accuracy of variant calls generated from WGS data.
Together, this work Is a shining example of how open science, fuelled by high-quality data and collaborative innovation, can lead to transformative advancements in clinical care.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in