Vernalis and Diamond turbocharge structure-based optimization of compounds
Published in Chemistry
There has been much interest in using Diamond’s XChem facility for high-throughput crystallographic fragment screening, to find compounds that bind to a protein. Recent examples include screening of 9 proteins from SARS-CoV-2 during the UK lockdown. Two of these campaigns, against Mpro and NSP3-macrodomain, are already made public through the Diamond website. Both demonstrated how, with a suitable crystal system, thousands of crystal structures can be determined in a few days, and fully modelled only weeks later.
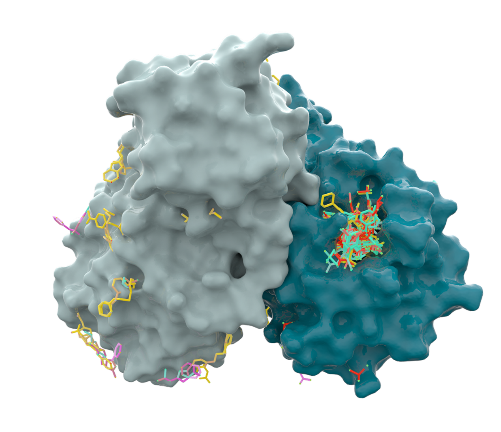
SARS-Cov Mpro dimer structure (grey and green) and all the fragments identified during the initial XChem screen [see preprint]
Such screens identify compounds that bind to specific sites on the protein crystal and confirm druggability of binding sites. However, as every fragment practitioner knows, identifying the fragment hits is the easy part: the real challenge is to elaborate and optimize the properties of the fragments into lead compounds with sufficiently high potency.
The process of compound optimization is time-consuming and reagent-hungry, requiring many cycles of synthesis of individual compounds followed by purification, characterization and making up solutions of defined concentration for measurement of binding affinity in some biochemical or biophysical assay. Recently, Vernalis scientists have developed an alternate approach to compound optimization which can work directly with crude reaction mixtures (CRMs). The binding of a ligand (L) to a protein (P) is usually written as a simple equilibrium:

where kon and koff are the rate constants for the binding and dissociation and which is related to the equilibrium dissociation constant KD (affinity).
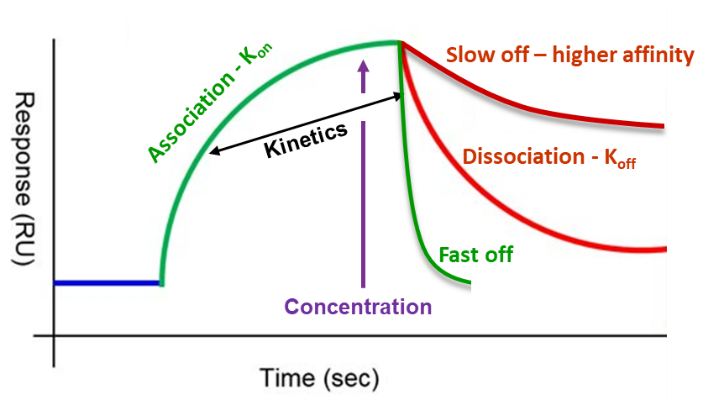
Although there can be changes to kon (e.g. conformational change), affinity improvements are usually from slower koff from additional interactions with the protein. koff is a first order rate constant, independent of [PL], so if there is some PL formed, then a slower koff is a surrogate for affinity improvement. Surface plasmon resonance (SPR) can rapidly identify changes in koff from flowing a CRM over a protein immobilized on the SPR sensor surface.
In this Communications Chemistry paper, through a very large set of experiments, Vernalis and XChem teams investigated thoroughly how high-throughput screening of such CRMs by crystallography can complement the off-rate experiments. A total of 83 CRMs (constructed from two different starting materials) were assessed against two related proteins, PDHK2 and Hsp90 in XChem and the results compared with off-rate screening by SPR. In total, more than 1500 structure determinations were made to establish the protocols, investigate reproducibility, and to compare manual and automated protocols for structure refinement – see the impressive Supplementary Table S1 of results. This was a massive effort by the lead crystallographer on the project, Lisa Baker, with great support from the team at Diamond, ironing out issues with the experimental and diffraction data analysis protocols.
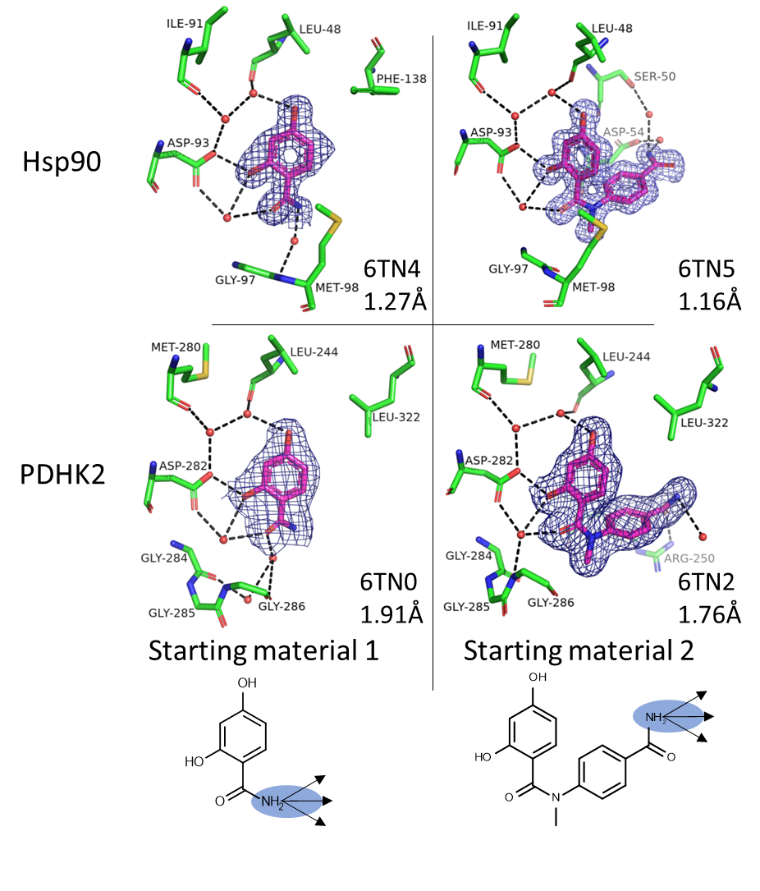
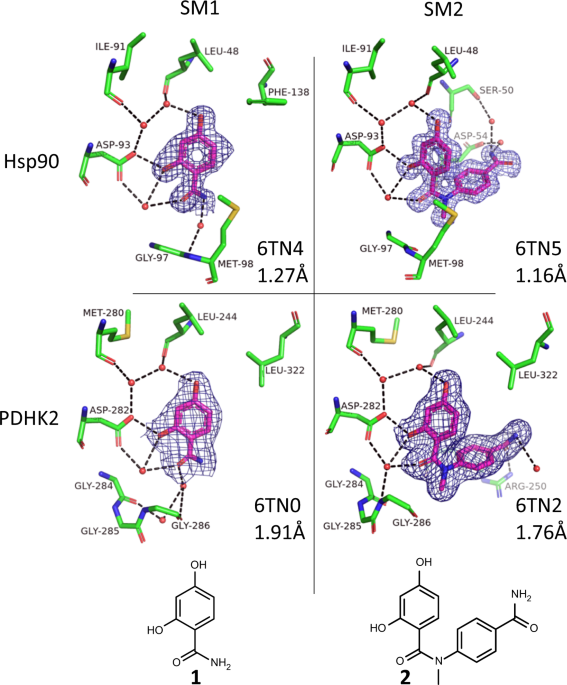
Electron density maps showing both starting materials bound to Hsp90 and PDHK2 protein crystals. The blue circles show the growing vector used for the synthesis of the CRMs.
The overall message is that most of the time (as for PDHK2), the results are consistent, and the protein crystal selects the higher affinity product from the CRM. What was exciting to see was that the crystal can also pick out the higher affinity enantiomer (Kd = 0.14 µM vs 17 µM) from the mixture in the CRM.
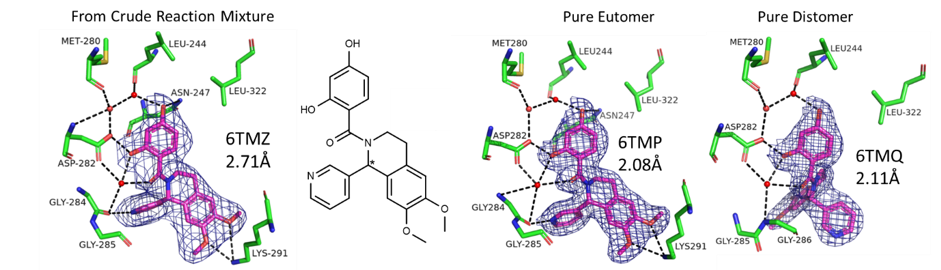
Electron density maps showing the higher affinity enantiomer in the CRM preferentially bound to the PDHK2 crystal structure. Separated, both enantiomers bind to the crystal structure.
For Hsp90, the results were less robust – probably because successful binding of a compound when soaked into a crystal is not just related to the affinity of binding as measured in solution. Taken together, the results suggest a protocol that can be followed for rapid optimization of fragments and hit compounds to higher affinity with structural information generated to guide further improvements.
You can find the the paper at https://doi.org/10.1038/s42004-020-00367-0
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Jun 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: May 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in