ViSNet: The universal molecular geometry modeling framework
Published in Chemistry, Cell & Molecular Biology, and Mathematics
Molecular modeling plays key roles in understanding bioactivity mechanisms, chemical property prediction, drug design and protein engineering. Geometry deep learning (GDL) is a widely used computational approach with low cost and high accuracy. Although there is huge progress over the past decade, there still exists some limitations to be solved: 1) Insufficient molecular interpretability: the deep neural networks act as a black box to make predictions but lack of deep insights into molecules; 2) Rapidly increasing computing costs as molecular size increases: the high-order Clebsch–Gordan products adopted in some SoTA approaches is computational intensive and thus blocks its applications for large molecules; 3) Lack of blind tests and evaluations in real applications: models are always tested on benchmark while the usefulness in real-world applications should be carefully evaluated.
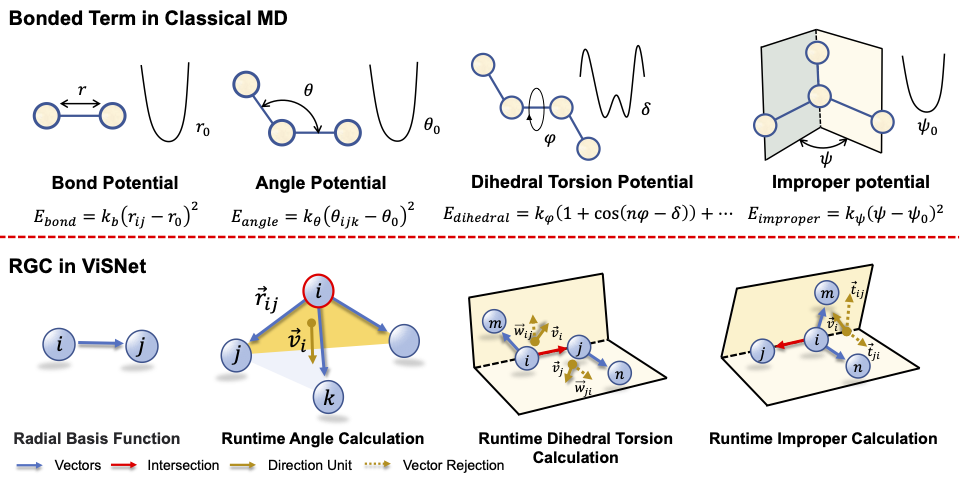
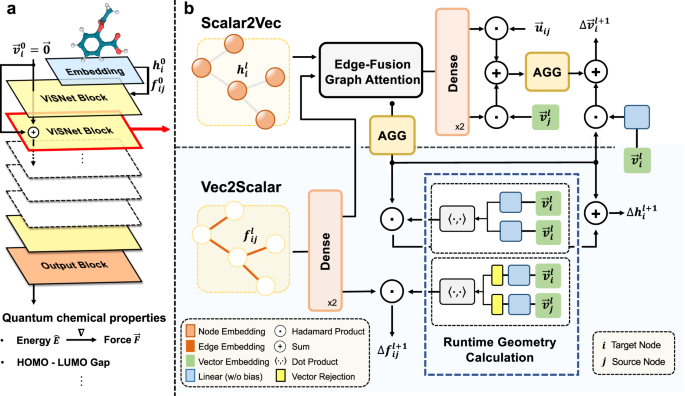
In light of this, we initially planned to design a model by making full of the domain knowledge of molecular structures efficiently. Classic molecular dynamics simulates molecular movements by explicitly describing bond length, bond angle and dihedrals in the potential energy function. Inspired by classic MD simulations, we convert such items into model design of ViSNet. Unlike directly adopting the angle or dihedral information by a simple feature engineering process, we proposed a concept "direction unit", the sum of all normalized vectors from the central atom to any of its first neighboring nodes, as a vectorized representation of the central node. We then designed Runtime Geometry Calculation (RGC) module to depict angles, dihedrals and so on as model operations. More importantly, the RGC calculations for both angles and dihedrals has only linear time complexity and together with vector-scalar interactive message passing mechanism (ViS-MP), it significantly accelerates the message passing process in molecular graph neural networks. The source code of ViSNet is available at https://github.com/microsoft/AI2BMD/tree/ViSNet .
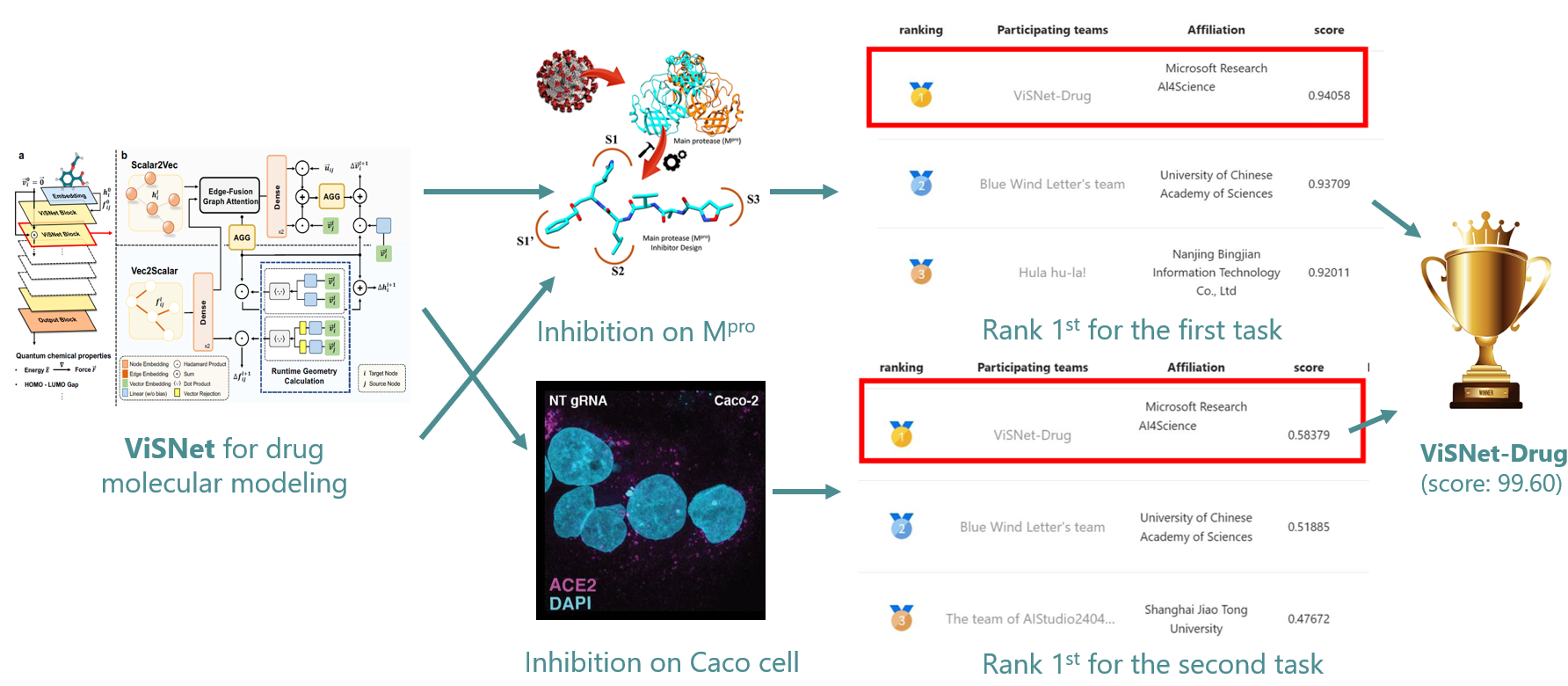
To examine the usefulness of ViSNet in real-world applications, we participated in the First Global AI Drug Development Competition. This competition is to predict the inhibitors against the main protease of SARS-CoV-2 given the sequence information (i.e., SMILES) of small molecules (https://aistudio.baidu.com/competition/detail/1012/0/leaderboard). There are 1,105 teams around the world participating in the competition, and ViSNet achieved the championship with a superior prediction accuracy. ViSNet takes the 2D structures of the molecules as inputs and predicts whether the molecules have inhibition ability to SARS-CoV-2 or not. The blind tests in the competition further consolidates the usefulness of ViSNet as a universal molecular geometry modeling framework in real-world applications.
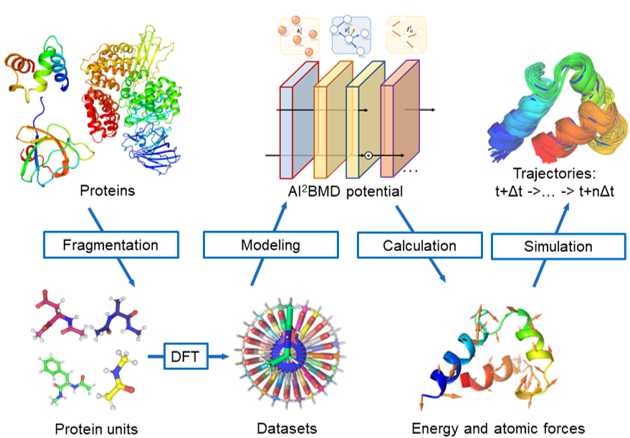
Furthermore, ViSNet is part of the outcomes of a larger project known as AI-powered Ab Initio Molecular Dynamics (AI2BMD) (https://microsoft.github.io/AI2BMD/index.html), which makes use of AI to do fast molecular dynamics simulation for large molecular systems with near ab initio accuracy. With ViSNet as the engine of molecular dynamics simulation, AI2BMD achieves near ab initio accuracy for energy and force calculations of proteins containing over 10,000 atoms. With the ability of simulating protein dynamics at ab initio accuracy, the project effectively complements laboratory experiments in understanding the dynamic aspects of various biochemical processes.
Dr. Tong Wang is the Senior Researcher at Microsoft Research AI4Science. He received his Ph.D at Tsinghua University and held a visiting Ph.D experience at Harvard University. His research focuses on algorithm design and applications on molecular dynamics simulation with artificial intelligence, machine learning force field, computer-aided drug discovery and protein structure prediction. Dr. Wang published a series of peer-reviewed papers as the first and corresponding authors on Nature, Nature Machine Intelligence, Nature Communications, Cell Research and so on, and holds several China and US patents. He is also the reviewers of Nature series journals and the honored reviewer of ACS publications. As the project lead, he proposed AI2BMD simulation program that transforms protein dynamics simulation with ab initio accuracy. As the team leader, he won the championship in the First Global AI Drug Development Competition (https://aistudio.baidu.com/competition/detail/1012/0/leaderboard) and the winner of OGB Large Scale Challenge@NIPS2022 competition. His research “Exploring the Regulatory Function of the N-terminal Domain of SARS-CoV-2 Spike Protein through Molecular Dynamics Simulation” won Top Downloaded Article Award by Wiley Press. He is the committee member of China Bioinformatics Society.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in