Visualizing nonequilibrium dynamics of macromolecular machines in atomic detail: an emerging paradigm that will innovate drug discovery
Published in Chemistry
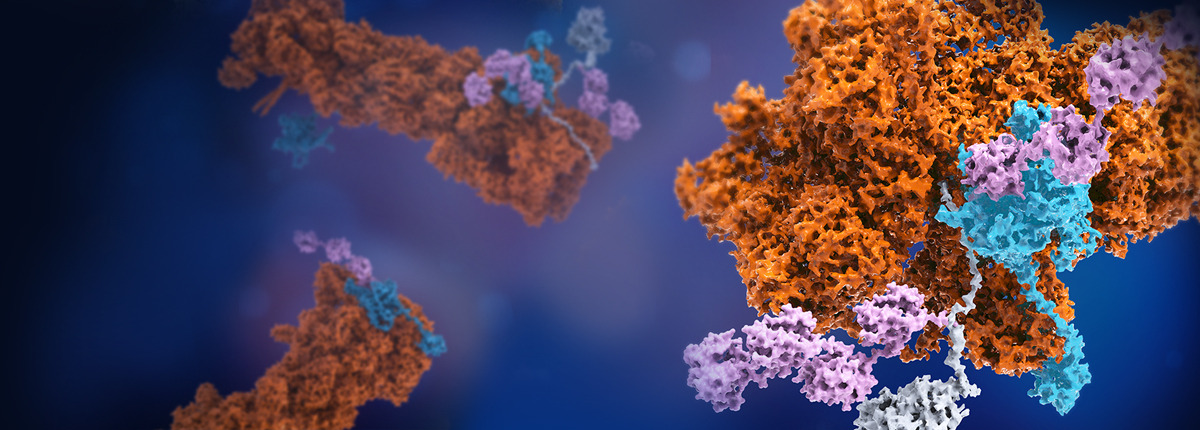

For a long time, solving a structure of a large macromolecular complex in an averaged conformation was the major efforts to understand the structure-function relationship of such a complex. In nearly all such structural studies, researchers often rely on several assumptions for simplifying structural determination. First, the macromolecular complex exists in a thermostable state, with all copies assuming identical conformation. Second, such a conformation informs on function of the complex or closely resembles the native state. In reality, both assumptions are not well held in that the complex in solution may experience conformational fluctuation and equilibrium thermodynamic sampling of multiple viable native conformations, if the energy landscape of the folded proteins is highly frustrated without domination of a single energy well. Once encountering its substrate or activated for catalysis, enzymatic complexes may enter nonequilibrium conformational transition for functional execution.
Visualizing nonequilibrium conformational dynamics of multi-protein holoenzymes at the atomic level have been largely inaccessible in the past. In such a process, many unstable and metastable intermediate conformations may be produced. These conformations of nonequilibrium intermediates are the real keys to understanding of how the chemical reactions and catalysis are executed. Unfortunately, their low abundance and extreme conformational heterogeneity make it very difficult to analyze. In the case of megadalton complex, it then becomes entirely impossible for existent technology to solve at the atomic level.
Time-resolved cryogenic electron microscopy (cryo-EM) has offered the possibility to attain this elusive goal, with several hurdles and limitations that must be solved before one can do so. First of all, a proper in vitro reconstruction of the biochemical reaction must be worked out and verified in complementary assays to sufficiently reproduce the functions of the systems in cells or in vivo. This can be a daunting task for many cellular biochemical processes that are insufficiently understood. With only one missing reactant or activator, the biochemical reactions may never take place in vitro. Second, cryo-EM reconstruction at high resolution still exercises image averaging from a large number of macromolecules of identical conformations. Image classification based on the 3D conformations, often referred to as 3D classification, is plagued by the poor signal-to-noise ratio (SNR) of raw cryo-EM images, which mostly falls in the range of 0.005-0.05 or even lower. This severely limits how many different 3D conformations can be reliably sorted out and reconstructed to high enough resolution. Current 3D classification methods utilizing maximum-likelihood estimation1,2 are capable of sorting out a few high-resolution conformations, often limited to 3-7 high-resolution conformers3-5. This approach suffices for “computational purification” of a couple of major conformations by discarding heterogeneous particle images via 3D classification. In one extreme case, when an exhaustive iteration of manually curated 3D classification was performed, up to 7 conformations have been solved to 3-3.6 Å resolution range, given more than 3.5-million single-particle images of substrate-bound 26S proteasome5. Recent development in mapping continuous motion of macromolecules from cryo-EM, without gauging 3D classification accuracy, allows visualization of conformational heterogeneity at moderate resolution but do not necessarily exhaust the search of the low-abundance conformers.
Owing to this major limitation, time-resolved cryo-EM has been limited to solve a few (2-3) conformational species after reactants are mixed. Reducing the requirement for image number while still achieving a resolution necessary for de novo modeling (1.0-3.6 Å range) remains a major challenge. This problem seems to be potentially tractable by integrating multiple machine learning methods in a synergistic manner6, and in several tested cases, allows significantly more conformers to be classified and refined to 3-4.9 Å range from the same dataset giving rise to 7 atomic structures of the 26S proteasome by conventional methods7. However, this new approach saves no computational costs and requires considerable reworking for the algorithmic restructuring for improving computing efficiency. In one case, this approach allowed us to truly advance time-resolved cryo-EM in solving 13 nonequilibrium conformers of the functioning proteasomes in the act of ubiquitylated substrate processing8, an intrinsically complex procedure, all in the resolution range of 3-3.6 Å.
On one hand, the field heralds the so-called “resolution revolution” for cryo-EM. On the other hand, the resolution of solving dynamic motion of macromolecules, particularly in the non-equilibrium process of enzymatic function against its substrate, remains moderate to low in most cases. Recent advance in the marriage of time-resolved cryo-EM and machine-learning-enhanced, high-accuracy 3D classification has provided us an example of how to tap into the conformational dynamics of important megadalton holoenzymes in actual nonequilibrium process of enzymatic actions on substrates. The demonstration of the feasibility of such experiments on the human 26S and 30S proteasome, which is truly as complex as ribosome and together governs the entire proteome homeostasis9, is necessary and will very likely inspire more experimental studies to follow. Going forward, several efforts must ensure to enable this emerging paradigm to thrive and prosper. First, the time-resolved cryo-EM sample preparation should be developed into a more principled protocol and technique, with higher reproducibility and best time-resolution available. Second, the ease of use and computing efficiency of the high-accuracy 3D classification software must be considerably improved to make it accessible to structural biologists in general. Third, the tools for fast atomic modeling must also be considerably enhanced and semi-automated, potentially benefiting from integrating with protein structure prediction tools like AlphaFold, in that the ability of reconstructing tens of high-resolution cryo-EM maps from a single dataset also drastically increases the workloads of atomic modeling.
More importantly, all these technical advance and improvement will eventually lead us to understand nonequilibrium dynamic processes of everything happening in cells atom by atom. Ultimately, a virtual cell could be built at the atomic level and all biochemical reactions can be accurately reproduced in silico, with the atomic-level knowledge we learned from time-resolved atomic visualization of all cellular biochemical reactions and macromolecular interactions. In the future, one could expect that all chemical compounds and molecular medicine constructs can be tested in a virtual cell in silico for drug discovery, the ultimate degree of virtual screen that might replace experimental screen completely. While it is hard to predict how long it will take to achieve this, it is certain that the journey to an augmented future has already started.
1 Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, https://doi.org/10.7554/eLife.42166 (2018).
2 Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290-296, https://doi.org/10.1038/nmeth.4169 (2017).
3 Chen, S. et al. Structural basis for dynamic regulation of the human 26S proteasome. Proc Natl Acad Sci U S A 113, 12991-12996, https://doi.org/10.1073/pnas.1614614113 (2016).
4 Zhu, Y. et al. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat Commun 9, 1360, https://doi.org/10.1038/s41467-018-03785-w (2018).
5 Dong, Y. et al. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49-55, https://doi.org/10.1038/s41586-018-0736-4 (2019).
6 Wu, Z. et al. Visualizing conformational space of functional biomolecular complexes by deep manifold learning. Int. J. Mol. Sci. 23, 8872. https://doi.org/10.3390/ijms23168872 (2022).
7 Wu, Z. et al. Hidden dynamics of proteasome autoregulation discovered by cryo-EM data-driven deep learning. bioRxiv, https://doi.org/10.1101/2020.12.22.423932 (2022).
8 Zhang, S., Zou, S., Yin, D. et al. USP14-regulated allostery of the human proteasome by time-resolved cryo-EM. Nature 605, 567–574, https://doi.org/10.1038/s41586-022-04671-8 (2022).
9 Mao, Y. Structure, Dynamics and Function of the 26S Proteasome. Subcell Biochem 96, 1-151, https://doi.org/10.1007/978-3-030-58971-4_1 (2021).
Biophysics and Chemical Biology
Follow the Topic
-
Nature
A weekly international journal publishing the finest peer-reviewed research in all fields of science and technology on the basis of its originality, importance, interdisciplinary interest, timeliness, accessibility, elegance and surprising conclusions.

Related Collections
With Collections, you can get published faster and increase your visibility.
Carbon Dioxide Removal
Publishing Model: Hybrid
Deadline: Jan 16, 2027



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in