When two proteomic platforms look at the same tumour sample, do they see the same biology?
Published in Cancer and Protocols & Methods
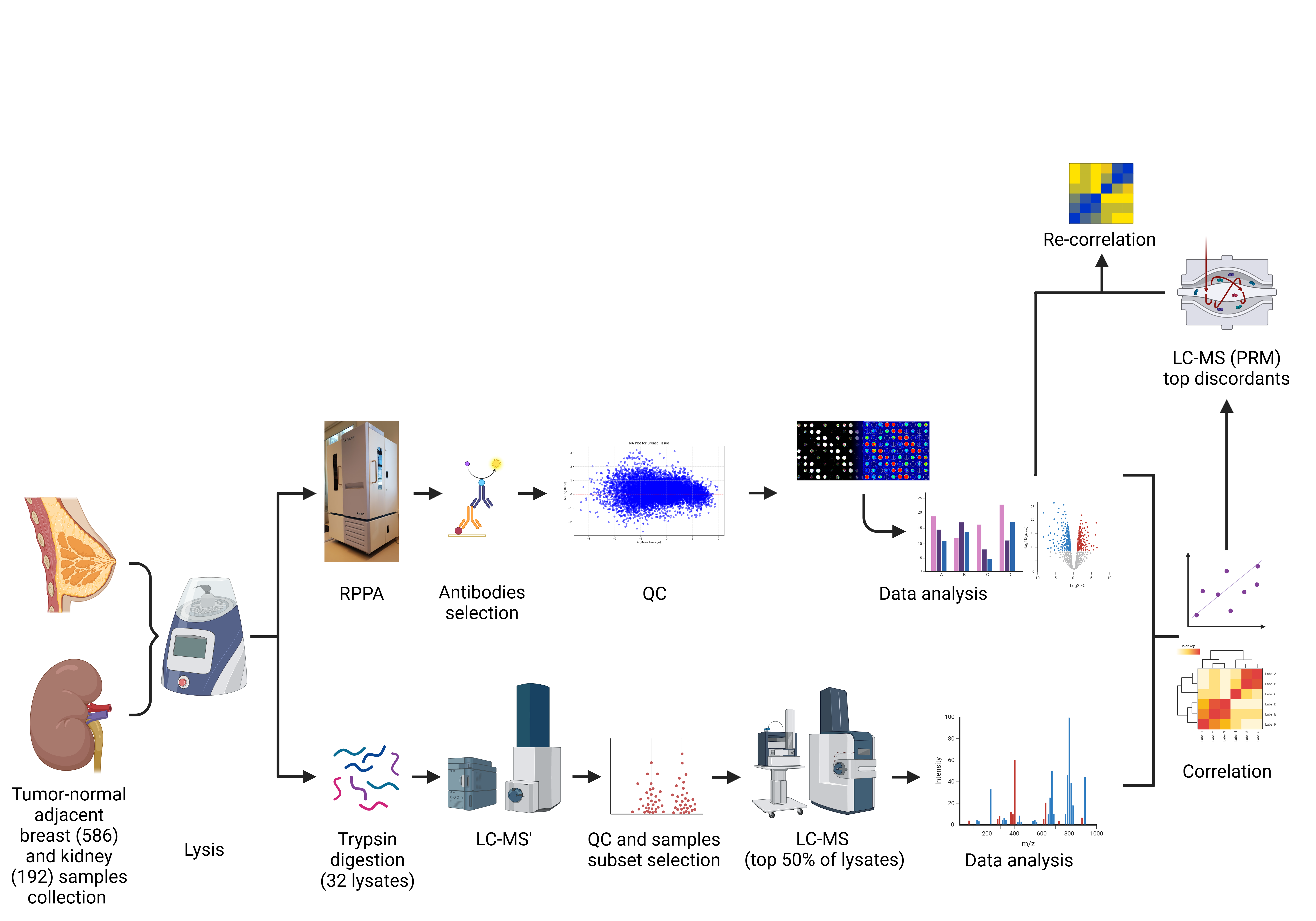

Mutations, copy-number changes and altered transcription can point us toward oncogenic mechanisms, yet they do not always predict protein abundance, protein state or pathway activity. For biomarker discovery and translational oncology, this creates a practical problem: which proteomic platform should we trust, and under what conditions?
In our study, we addressed this question by comparing two widely used but analytically distinct proteomic technologies: Reverse Phase Protein Array (RPPA), and liquid chromatography–mass spectrometry (LC-MS). Rather than treating them as competing methods, we asked a narrower and more clinically relevant question: when both platforms measure cancer-relevant proteins from the same lysates, how often do their tumour-versus-adjacent-normal readouts agree?
We focused on matched tumour and adjacent normal tissues from breast and kidney cancer patients. The scale of the RPPA component was substantial: 586 breast tissue samples, representing 293 matched pairs, and 192 kidney tissue samples, representing 96 matched pairs. Samples were collected, barcoded, frozen and processed into lysates for high-throughput RPPA profiling (https://rppa.hu/). The samples database is handled by the Laboratory Information system of the Budapest Reverse Phase Protein Array facility, (LIBRA), which supports the structured management of the sample collection and associated workflow information.
The protein panel was anchored to the Ion AmpliSeq Cancer Hotspot Panel v2 gene list. This choice was deliberate. These are genes commonly discussed in cancer genetics, but our purpose was not to detect mutations. Instead, we asked whether proteins encoded by these cancer-relevant genes could be quantified reproducibly and comparably across proteomic platforms. After extensive antibody screening, RPPA-suitable antibodies were identified for 48 of the 50 proteins in the panel, complemented by a set of cellular markers representing different cellular functions and compartments.
Each lysate was printed as a five-point dilution series, with multiple technical replicates, generating 40 technical spots per sample per antibody-stained slide.
RPPA provided the high-throughput backbone of the study. It allowed us to quantify selected proteins across hundreds of matched tumour and adjacent-normal samples using minimal material. LC-MS, by contrast, was applied to a smaller subset because of its much greater cost, analytical complexity and sample-throughput constraints. We used a tiered LC-MS design: an initial data-dependent acquisition screen on 32 lysates, followed by deeper data-independent acquisition and targeted parallel reaction monitoring (PRM), on 16 selected lysates from four matched breast and four matched kidney tumour-adjacent pairs. Crucially, all cross-platform comparisons used aliquots from the same original lysates, avoiding a common confounder in method-comparison studies: comparing technically different preparations of biologically heterogeneous tissues.
The first observation was that RPPA captured protein-specific tumour-normal differences, but not a simple universal pattern. In breast samples, MET showed a clear cohort-level decrease among the hotspot-panel proteins, while CAV1 showed a marked decrease among cell markers. In kidney samples, the α subunit of Na⁺/K⁺ ATPase, NAKAa1, showed the clearest cohort-level decrease among the cell markers. However, only a limited subset of proteins reached significance after multiple-testing correction. The differences were therefore protein-specific, not evidence for broad organ-wide up- or down-regulation.
We also examined whether normalizing RPPA values to cellular markers could reduce dispersion. Vimentin, VIM, emerged as a useful empirical normalizer in this dataset, but we interpret this cautiously. It was a practical, dataset-specific choice rather than a universal recommendation. This point matters because normalization decisions can materially affect cross-platform correlations, especially in bulk tumour tissues where epithelial, stromal, immune and vascular components may vary between samples.
The central result of the study was the cross-platform comparison. RPPA and LC-MS agreed for some proteins, but agreement was partial and strongly protein dependent. Some targets showed concordant tumour-to-normal fold changes across platforms. Others showed weak correlations, no clear relationship, or even opposite-direction changes. Targeted PRM improved agreement for selected discordant proteins in some cases, but it did not eliminate the discrepancies. In other words, RPPA and LC-MS were not interchangeable for all proteins in the panel.
This is not surprising when one considers what each platform measures. RPPA measures antibody recognition of a protein epitope in a lysate. Its strengths are sensitivity, throughput and efficiency, but its accuracy depends on antibody specificity, epitope accessibility and the availability of high-quality antibodies. LC-MS infers protein abundance from detected peptides after proteolytic digestion. It is antibody-independent and can, in principle, cover far more of the proteome, but it is affected by peptide detectability, ionization efficiency, missing values, dynamic range limitations and spectral interference. A protein may be clearly visible to an antibody but poorly represented by proteotypic peptides, or vice versa.
Mutations and post-translational modifications add further complexity. A tumour-specific mutation may alter an antibody epitope, affect peptide detection, or be missed by a standard reference-database MS search. Similarly, phosphorylation, ubiquitination, acetylation or glycosylation can alter antibody binding, proteolytic cleavage or peptide ionization. Thus, discordance between RPPA and LC-MS should not automatically be interpreted as biological insight. In our view, it is better treated as an analytical warning signal.
This leads to the main conceptual conclusion of the study: orthogonal proteomics is most useful here as a confirmatory strategy. When RPPA and LC-MS converge on the same direction of change in the same lysates, confidence in that candidate biomarker increases. When they diverge, the target should be considered unresolved until examined by additional targeted assays, larger cohorts, proteogenomic information or clinically validated methods. This is particularly relevant during biomarker discovery, assay development and early translational validation. It does not imply that routine clinical decision-making should require repeated orthogonal testing once an assay has already been analytically validated.
The study also has limitations. The samples were not sequenced for mutations in the respective genes. We did not have detailed clinical histories, tumour grading, molecular subtyping or tumour-purity estimates. The LC-MS validation cohort was necessarily small, with four matched tumour-normal pairs per tissue type in the deeper DIA/PRM analyses. The RPPA values were derived from multi-step processing of dilution-series data, and formal propagation of all upstream uncertainty was not performed. These points restrict how strongly individual protein correlations should be interpreted.
Even with these limitations, the message is clear. In cancer proteomics, platform choice is not a neutral technical detail. RPPA and LC-MS each provide valuable information, but they do not always provide the same information. For translational research, this is not a weakness; it is a design consideration. A robust biomarker pipeline should use cross-platform agreement to prioritize candidates and cross-platform disagreement to identify proteins requiring further scrutiny.
The broader implication is that precision oncology will benefit from proteomic workflows that are both technically rigorous and analytically modest. No single platform captures the full complexity of the tumour proteome. The more useful question is not which method is universally superior, but how each method can be deployed at the right stage: RPPA for scalable, targeted profiling; LC-MS for broader and antibody-independent discovery; and targeted MS or other orthogonal assays for focused validation. Our study provides a same-lysate benchmark for making those decisions more explicitly.
Follow the Topic
-
Scientific Reports
An open access journal publishing original research from across all areas of the natural sciences, psychology, medicine and engineering.

Related Collections
With Collections, you can get published faster and increase your visibility.
Infectious disease diagnostics
Publishing Model: Open Access
Deadline: Sep 23, 2026
AI in Education
Publishing Model: Open Access
Deadline: Oct 09, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in