A leap on protein isoform quantification in shotgun proteomics
Published in Protocols & Methods

The quantification of the abundance of all proteins in biological samples has long been a dream of biologists. Shotgun proteomics is widely used for protein identification and quantification in biomedical research, but protein isoform characterization is challenging due to the extensive number of peptides shared across proteins, hindering our understanding of protein isoform regulation and their roles in normal and disease biology. Several approaches have been proposed to allocate these degenerate peptides for protein-level quantification1-3. Among them, the parsimony-based approach4 was widely used and integrated into many popular proteomics data analysis tools, including MaxQuant5 and FragPipe6. In general, this approach collapses proteins with the same set of supporting peptides together with those that are supported by a subset of these peptides into a protein group7. For protein quantification, peptides shared by multiple protein groups are either ignored or assigned to the group with the largest number of associated peptides. Typically, one representative protein (i.e., the one with the largest number of associated peptides) is selected from each protein group for reporting.
In the early stages of proteomics, only thousands of peptides can be identified for a run. The parsimony-based approach played a critical role in preventing overstating the number of proteins in protein inference. However, with the improvement of the LC-MS/MS technology, the bottom-up proteomics approach can identify up to one hundred thousand of peptides in a single run. This approach may limit the potential for protein isoform characterization. First, protein isoforms without uniquely identified peptides are essentially ignored. Secondly, the assignment of shared peptides to the protein groups and proteins with the largest number of associated peptides for quantification may not necessarily be the correct solution. In this study, we systematically assessed above issues of parsimony-based approach using in silico digestion of the Refseq protein database (Fig. 1). Among the 588,545 resulting peptide sequences, 2.8% could be associated with multiple genes, 13.6% to genes with a single protein isoform, and 83.5% to genes with more than one isoform. Within the group of multi-isoform peptides, around half could be mapped to all protein isoforms of a gene, and thus providing no information for isoform discrimination; however, another half, or 246,615 peptides, could be uniquely mapped to one isoform or a subset of isoforms. Moreover, peptide distributions from real experiments are similar to that of in silico digestion. These data suggest an urgent need for computational methods that can effectively utilize peptides mapped to multiple genes, a single isoform or a subset of isoforms.
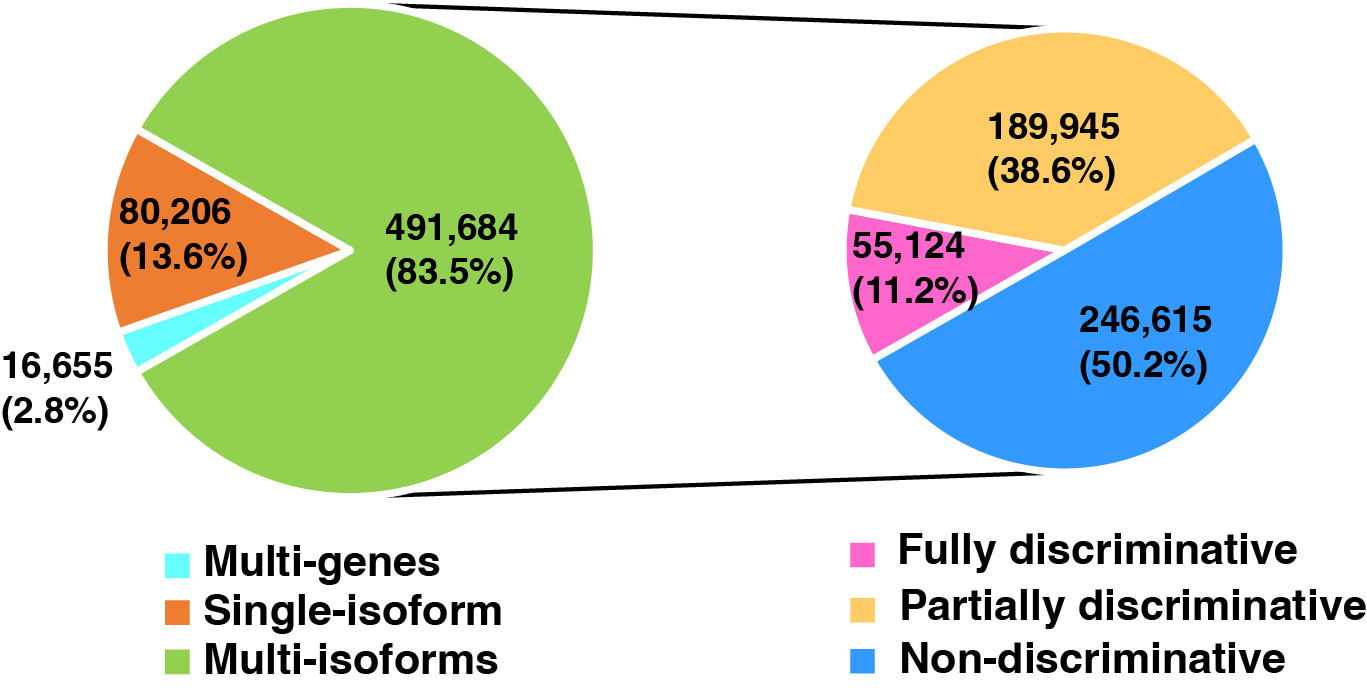
Figure 1: Classification of the in silico digested peptides based on their mapping to genes and protein isoforms.
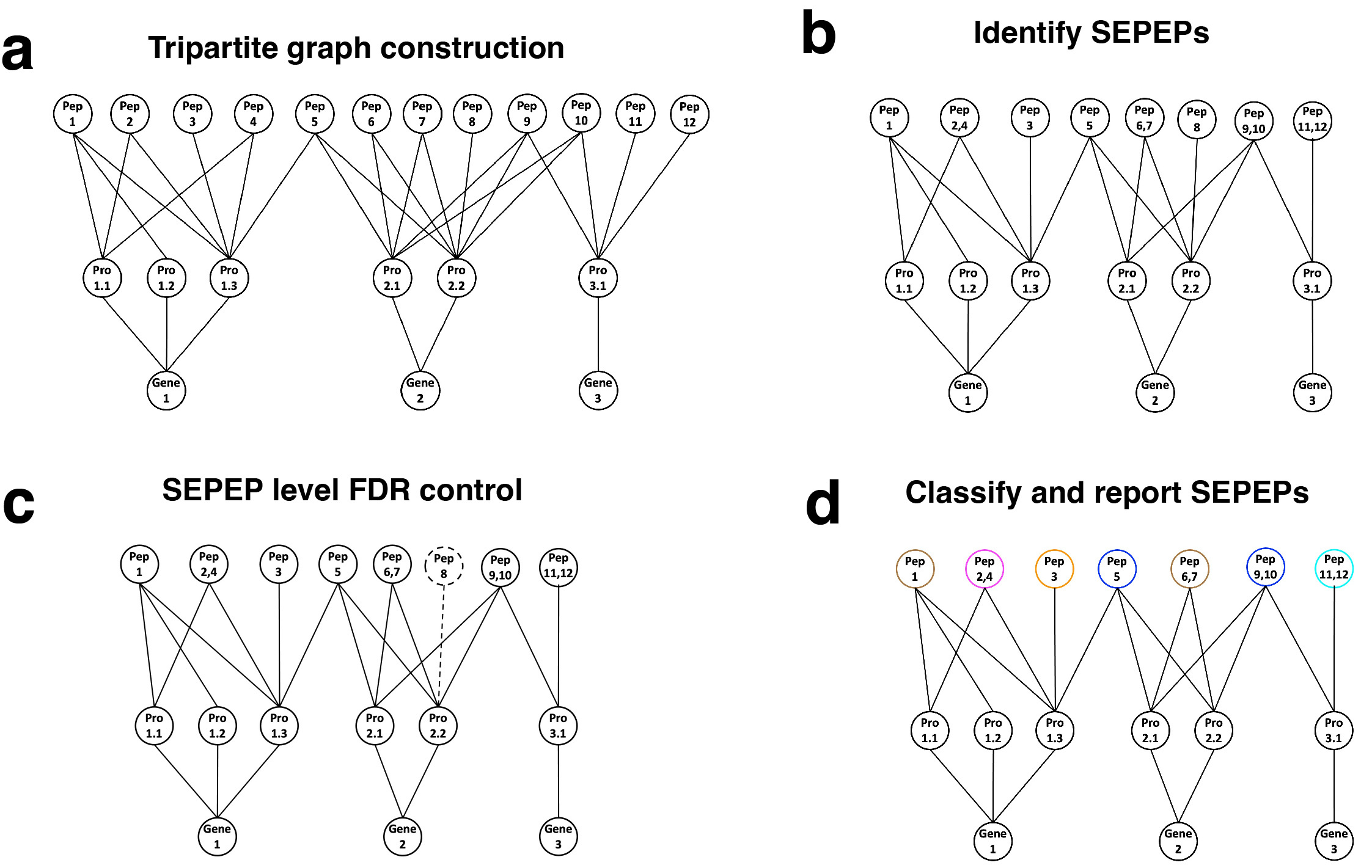
To achieve this objective, we proposed a tripartite graph modeling approach to represent the above peptides more accurately. First, a tripartite graph is built with three sets of vertices representing all peptides identified in a study (Pep1-Pep12), proteins to which the peptides can be mapped (Pro1.1-Pro3.1), and host genes of the proteins (Gene1-Gene3), respectively, and the vertices are connected by edges indicating their mapping relationships (Fig. 2a). Compared to the bipartite graph used in the parsimony-based approach4, peptides mapped to multiple genes will be kept in the tripartite graph. Second, peptides connected to the same set of protein vertices are grouped together and defined as a group of structurally equivalent peptides (SEPEP), leading to eight SEPEPs in Fig. 2b. Third, the target-decoy approach is used to estimate false discovery rate (FDR) at the SEPEP level. SEPEPs with FDR >0.01 are excluded from further analysis (Fig. 2c). Finally, the remaining SEPEPs are classified into five classes based on their patterns of connections to source proteins and genes in the tripartite graph (Fig. 2d). Class 1 through 5 correspond to single isoform SEPEPs, fully discriminative SEPEPs, partially discriminative SEPEPs, non-discriminative SEPEPs, and multi-gene SEPEPs, respectively.
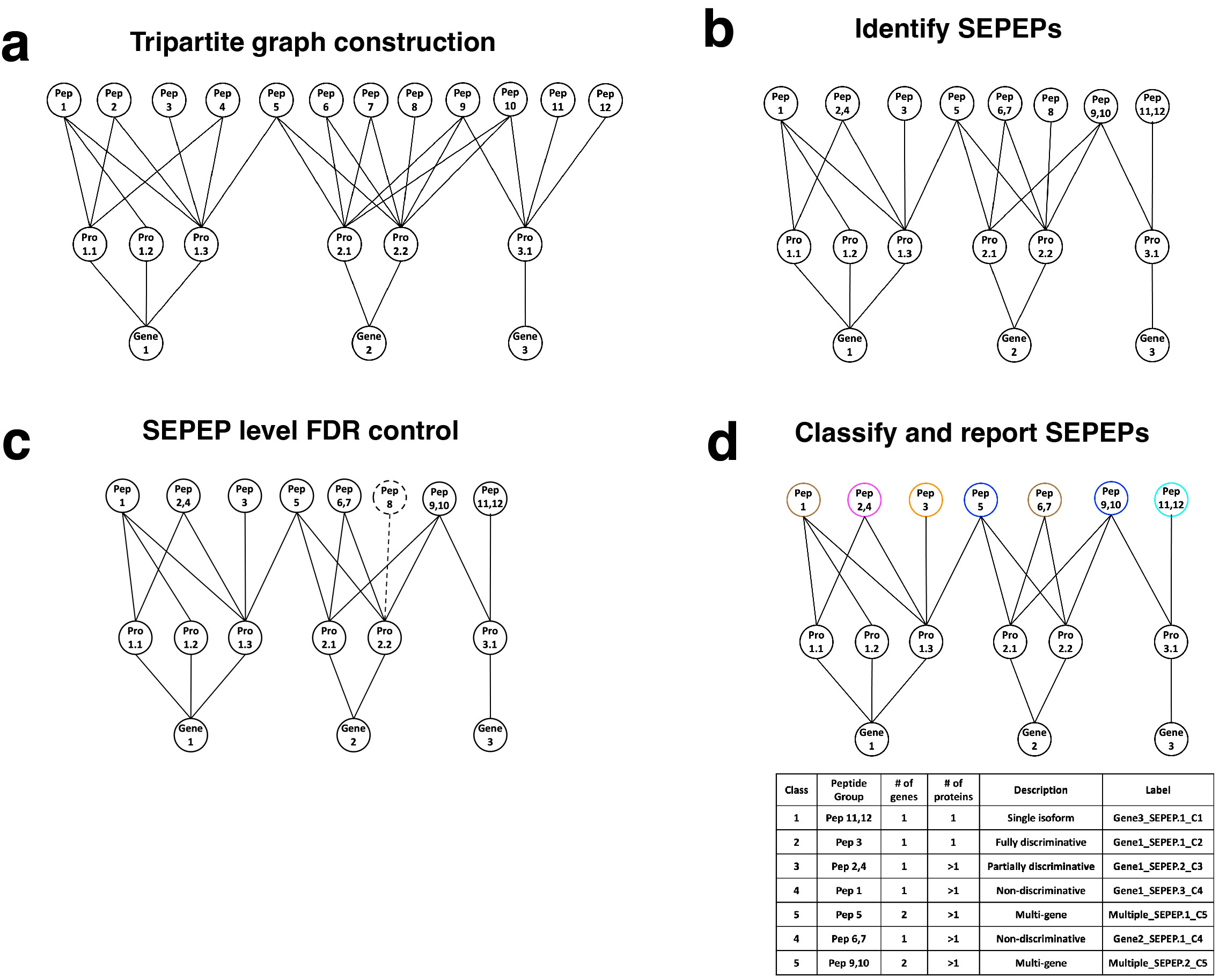
Figure 2: Overview of the tripartite graph modeling approach. a Tripartite graph construction: connect peptides to all proteins that contain them, and proteins to the host genes, to form a tripartite graph. b SEPEP identification: identify and group structurally equivalent peptides (SEPEP), i.e., peptides connecting to the same set of proteins in the graph. c FDR control: calculate SEPEP level FDR and remove SEPEPs with an FDR higher than the pre-specified threshold, e.g., FDR > 0.01. d SEPEP classification and reporting: classify SEPEPs based on their patterns of connections and report SEPEP level quantification.
Evaluated on three public data sets8-10, SEPepQuant reported thousands of fully discriminative, partially discriminative, and multi-gene SEPEPs. It can identify up to 34 times more genes with multiple features compared to parsimony model-based methods. For example, SEPepQuant confirmed a mutually exclusive spliced exons (MXE) event in TPM1 in an iPSC data set8. This MXE event was found by searching a junction peptide only database in the original iPSC publication. Applying SEPepQuant to a liver cancer data set9, we identified 18 potential isoform level regulation events that are associated with tumor initiation and progression. We confirmed a SLK exon skip event, which was upregulated in tumor and was associated with worse survival, by parallel reaction monitoring. In summary, SEPepQuant presents a leap in protein isoform quantification in shotgun proteomics. It has broad applications to biological and translational research.
To find more about SEPepQuant, the full article can be found here (https://www.nature.com/articles/s41467-023-41558-2). SEPepQuant is freely available on GitHub: https://github.com/bzhanglab/SEPepQuant.
Reference:
1 Gerster, S. et al. Statistical approach to protein quantification. Mol. Cell. Proteomics 13, 666–677 (2014).
2 Forshed, J. et al. Enhanced information output from shotgun proteomics data by protein quantification and peptide quality control (PQPQ). Mol. Cell. Proteomics 10, M111.010264 (2011).
3 Dermit, M., Peters-Clarke, T. M., Shishkova, E. & Meyer, J. G. Peptide Correlation Analysis (PeCorA) Reveals Differential Proteoform Regulation. J. Proteome Res. 20, 1972–1980 (2021).
4 Zhang, B., Chambers, M. C. & Tabb, D. L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557 (2007).
5 Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
6 da Veiga Leprevost, F. et al. Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870 (2020).
7 Zhang, B., Chambers, M. C. & Tabb, D. L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557 (2007).
8 Lau, E. et al. Splice-Junction-Based Mapping of Alternative Isoforms in the Human Proteome. Cell Rep. 29, 3751–3765.e5 (2019).
9 Gao, Q. et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 179, 561–577.e22 (2019).
10 Jiang, Y. et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257–261 (2019).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in