The synthesis of chiral compounds with high enantioselectivity is vital for drug development. N-oxides, featuring a stereogenic nitrogen center, are prominent in natural products and play crucial roles as ligand scaffolds in organometallic chemistry. They also serve as key intermediates in pharmaceuticals and agrochemicals. However, achieving the direct asymmetric oxidation of amines to chiral N-oxides with high enantioselectivity has been challenging due to the rapid inversion of the nitrogen atom’s lone pair, often resulting in racemization.
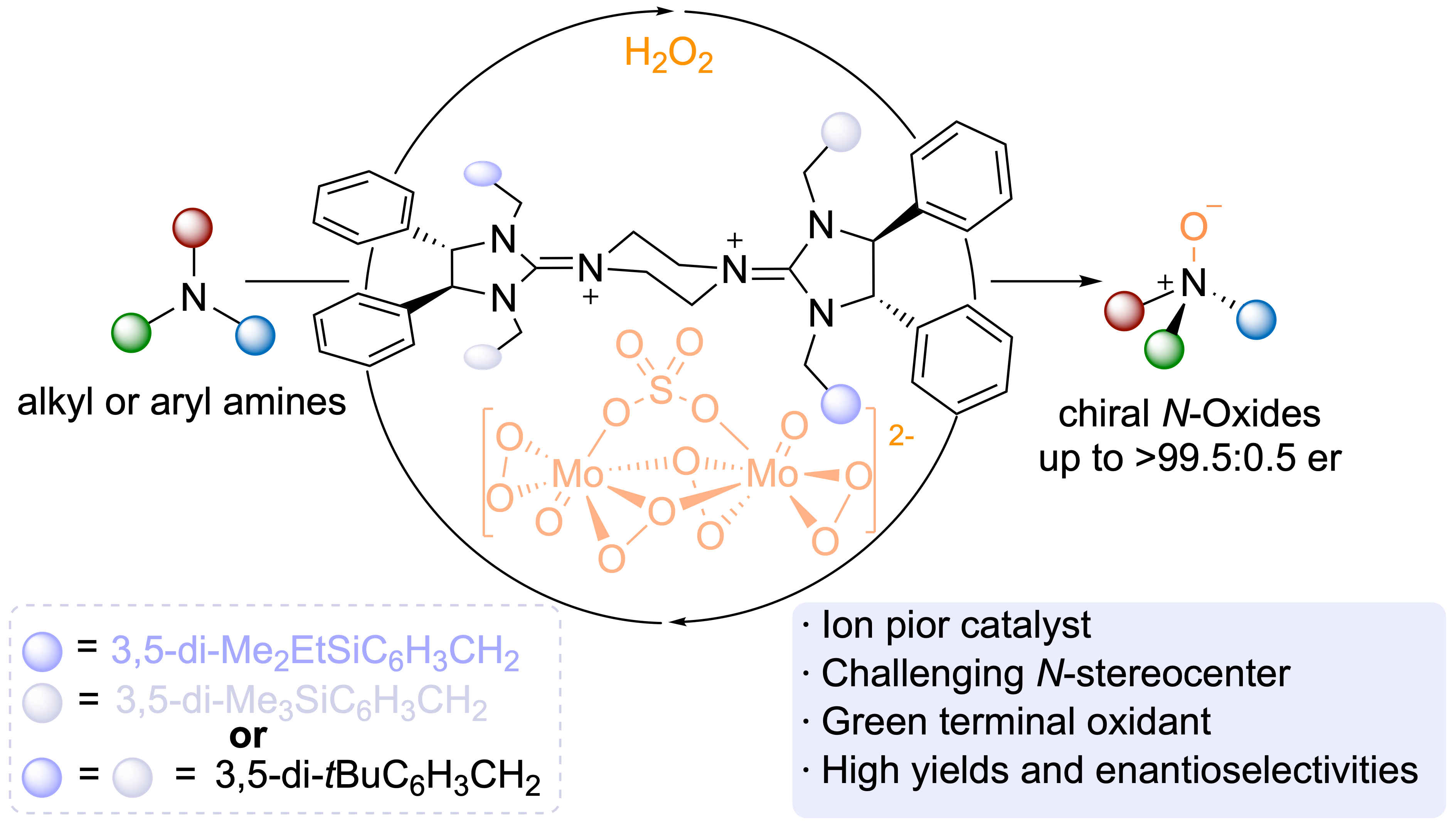
A recent breakthrough introduces a novel ion-pair catalyst that significantly advances the field of asymmetric N-oxidation. This catalyst, composed of a chiral bisguanidinium [BG]²⁺ cation paired with an achiral oxodiperoxomolybdosulfate anion, has demonstrated exceptional efficiency (Figure 1). X-ray crystallography revealed that a silyl group substituent on the cation plays a critical role in controlling the chiral environment around the amine substrate, enabling highly efficient and enantioselective oxidation. This innovative method has been validated on a gram scale and applied to the synthesis of pharmaceutical analogues, including a direct synthesis of a trimetazine antihistamine analogue.

Figure 1. Ion-pair catalyzed asymmetric oxidation
Mechanistic studies provided deeper insights into the process. Only one of the two peroxo groups in the oxodiperoxomolybdosulfate anion is involved in the oxidation, with an oxygen atom exchange occurring between the product and the substrate. Initially, this results in a low enantiomeric excess (ee) of the product, which increases as the reaction proceeds. Density Functional Theory (DFT) calculations modelled the key transition state, revealing the critical interactions between the catalyst and the substrate (Figure 2).
The proposed mechanism suggests that, upon activation by hydrogen peroxide, silver molybdate and potassium bisulfate generate the oxodiperoxomolybdosulfate anion in the aqueous phase. This anion is then captured by the bisguanidinium cation in the organic phase, where it participates in the asymmetric oxidation of the substrate. The reduced anion returns to the aqueous phase, where it is re-oxidized, completing the catalytic cycle.
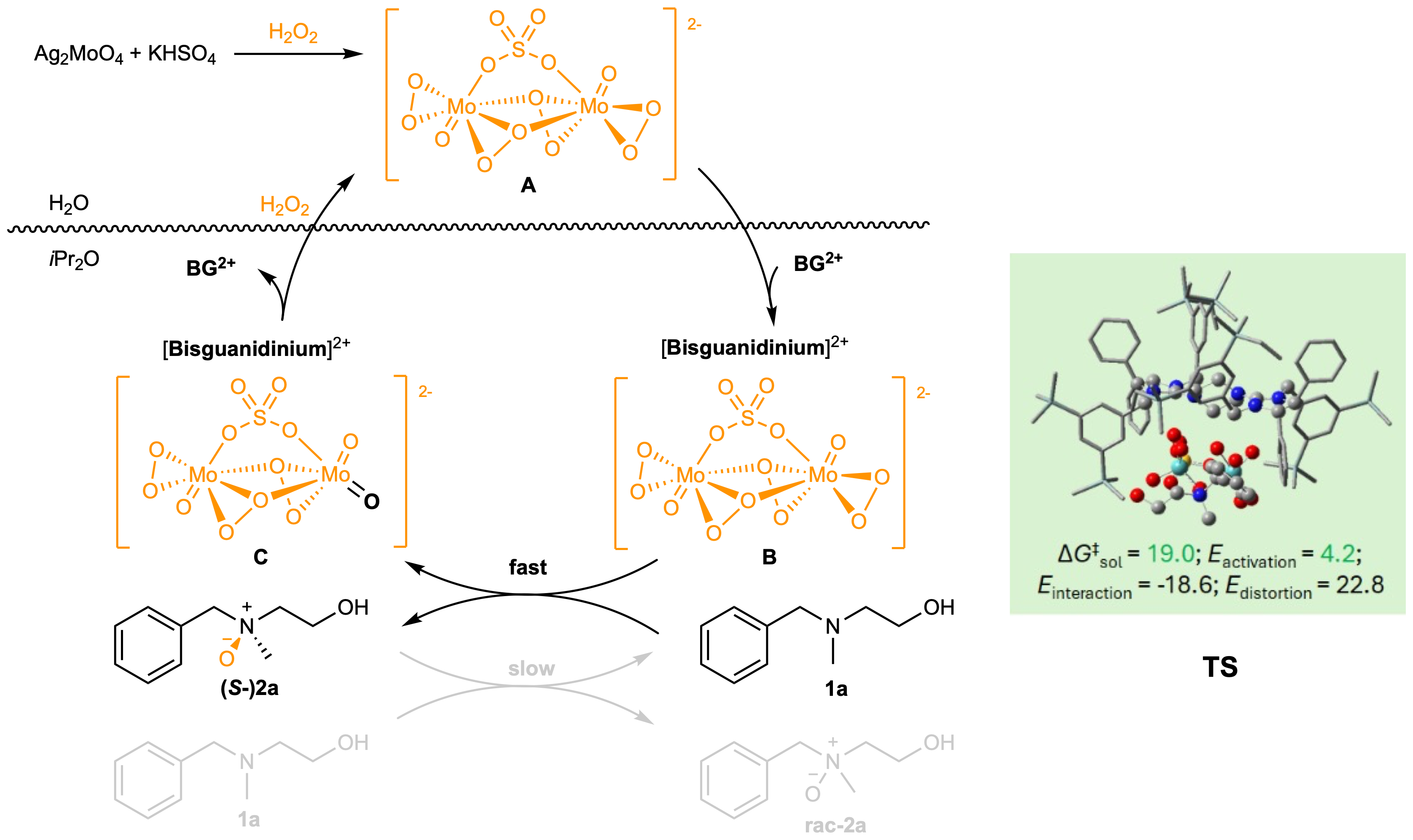
Figure 2. Working model and transition state
This research not only introduces a groundbreaking catalyst for the enantioselective synthesis of N-oxides but also provides a roadmap for designing future catalysts. The method’s high enantioselectivity, scalability, and applicability to pharmaceutical synthesis represent a significant advancement in asymmetric catalysis, opening new avenues for the development of chiral N-oxides in drug discovery and beyond.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Jun 01, 2026
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in