Behind the paper: DestVI identifies continuums of cell types in spatial transcriptomics data
Published in Bioengineering & Biotechnology
Explore the Research
DestVI identifies continuums of cell types in spatial transcriptomics data - Nature Biotechnology
DestVI models continuous cell states in spatial transcriptomics data.
Spatial transcriptomics (ST) is a technique for evaluating gene expression in-situ while recording the location from which RNA molecules are captured while maintaining high spatial resolution. Named 2020 Methods of the Year, ST comprises a set of technologies for characterizing the heterogeneity of cellular states and their organization in niches at the tissue level. The experimental assays evolve and improve quickly, but all remain limited in several types of resolution such as the number of genes that can be captured, or the spatial resolution (from dozens of cells down to subcellular fields of view). Consequently, there remain many open questions as to which biological phenomena may benefit from these technologies, but also as to what are the most helpful statistical tools that one can build to gain insight from these data. Our team was unanimously interested in both.
I (Romain) had worked on several methodology projects as a graduate student in Nir Yosef’s lab, including the development of single-cell Variational Inference (scVI) -- a probabilistic framework for analyzing single-cell RNA sequencing data. Initially, I was interested in ST data analysis from the perspective of modeling both modalities to recognize spatial gene expression patterns. I aimed at building an automated pipeline for the discovery of spatially-variable, interpretable gene programs. After months of drawing models, I felt that I needed some insights from analyzing real data in order to formulate a machine learning problem I would find exciting but also biologically relevant.
Baoguo and Hadas from Ido Amit’s lab wanted to benchmark 10x Visium as a method, to verify whether it can effectively localize cell state heterogeneity derived from single-cell RNA sequencing. Indeed, each capture spot of the 10x Visium assay may contain up to dozens of cells, which makes spatial identification of rare cell populations potentially challenging. Baoguo is an expert in microscopy imaging and could validate any further findings. For their benchmarking study, they had in mind two biological systems that were profiled previously in their lab (Figure 1). First, the comparative study of murine lymph nodes after exposure to mycobacteria compared to a control. This is an important and valuable example, as the tissue is well structured and we expected a cellular and molecular response consistent with the previous profiling done using scRNA-seq. Potentially, this choice promises to not only test the data but also the computational methods, as to whether they can identify the expected response. Second, we studied a solid tumor mouse model, for which we had much less prior biological information about the organization into compartments. This experiment, in contrast to the lymph node, was thought to be akin to a case study.
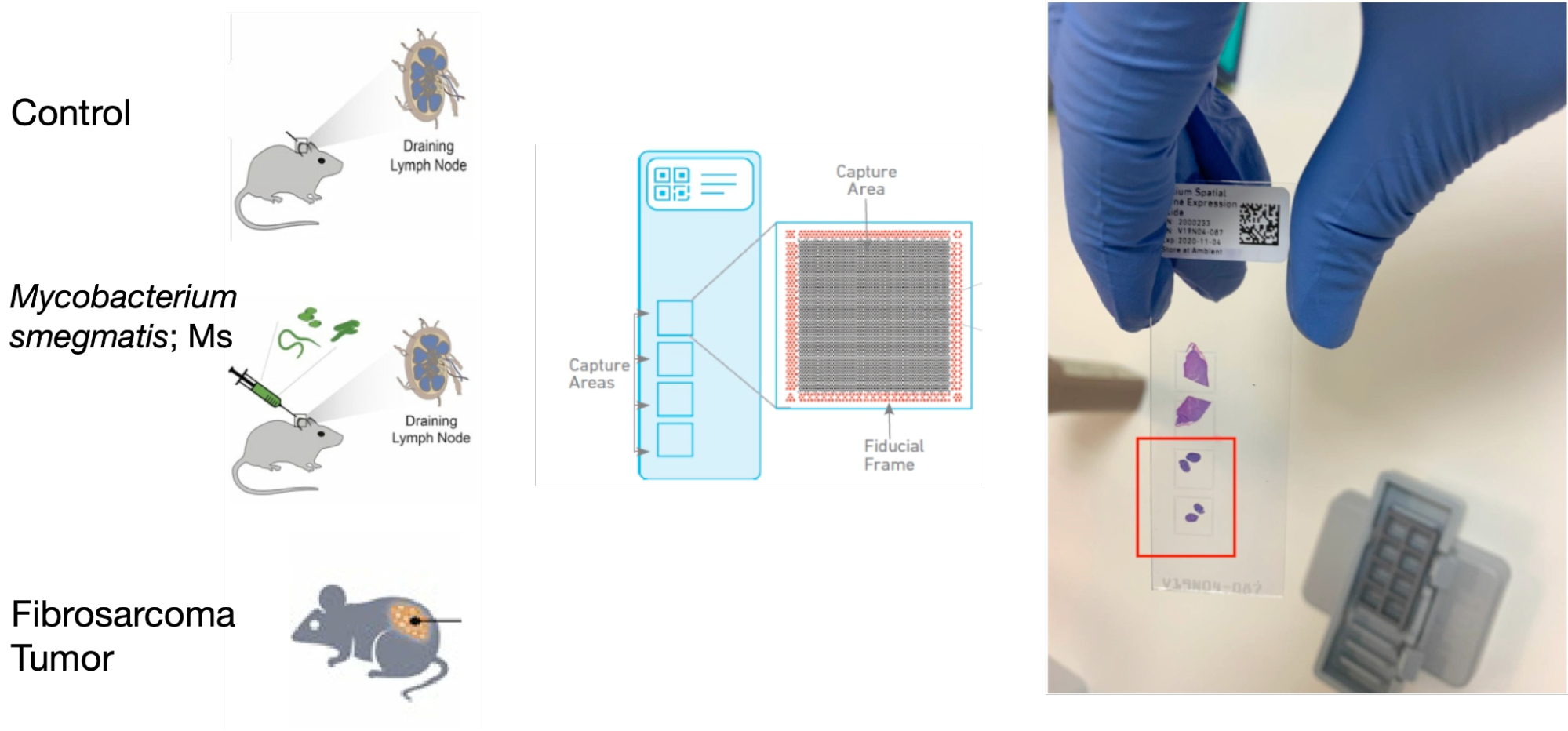
Figure 1: Our Visium experimental design. The red square indicates the capture areas used for the murine lymph nodes.
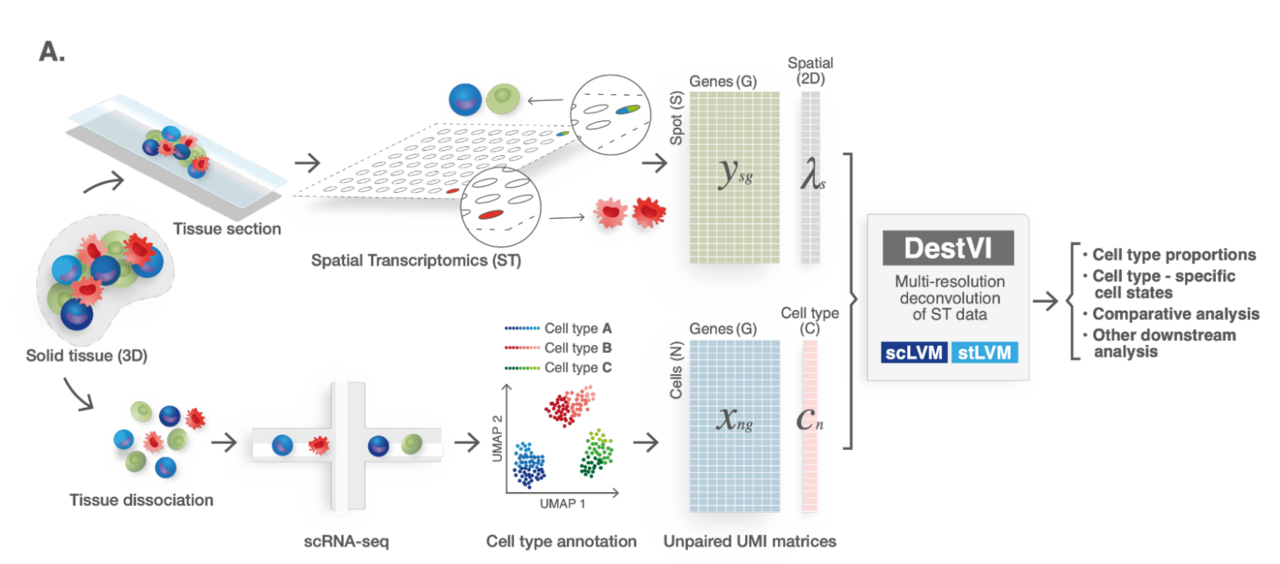
Us joining efforts marked the beginning of a wonderful collaboration. I started processing the data after Baoguo and Hadas profiled the tissues using 10x Visium. I annotated the single-cell data using my previous methodology (scVI & single-cell ANnotation with Variational Inference; scANVI) and then applied several existing methods for mapping cell types onto the spatial coordinates of the tissue (such as Stereoscope). These methods consist in clustering the scRNA-seq data to a certain resolution (discrete cell states), and then estimating the proportion of cells from every (discrete) state in every spot in the spatial data (our method has identical inputs; see Figure 2). Interestingly, the existing methods reasonably stratified the lymph node into compartments (T cells region, B cells region) but could not identify the phenotype characterized in the previous scRNA-seq work (interferon signaling in monocytes after exposure to the mycobacteria). The leading hypothesis explaining this failure was that monocytes are a rare population in the scRNA-seq data, therefore the sequential clustering of the data may lead to sample sizes that are too low for proper estimation. From a modeling perspective, we were looking at a critical computational problem: how can we describe cells as both belonging to discrete types (monocytes, T cells etc.) and to states that are continuous in nature (e.g., activation of T cells)? Then, how to map those states onto the spatial data, and interpret them as gene programs.
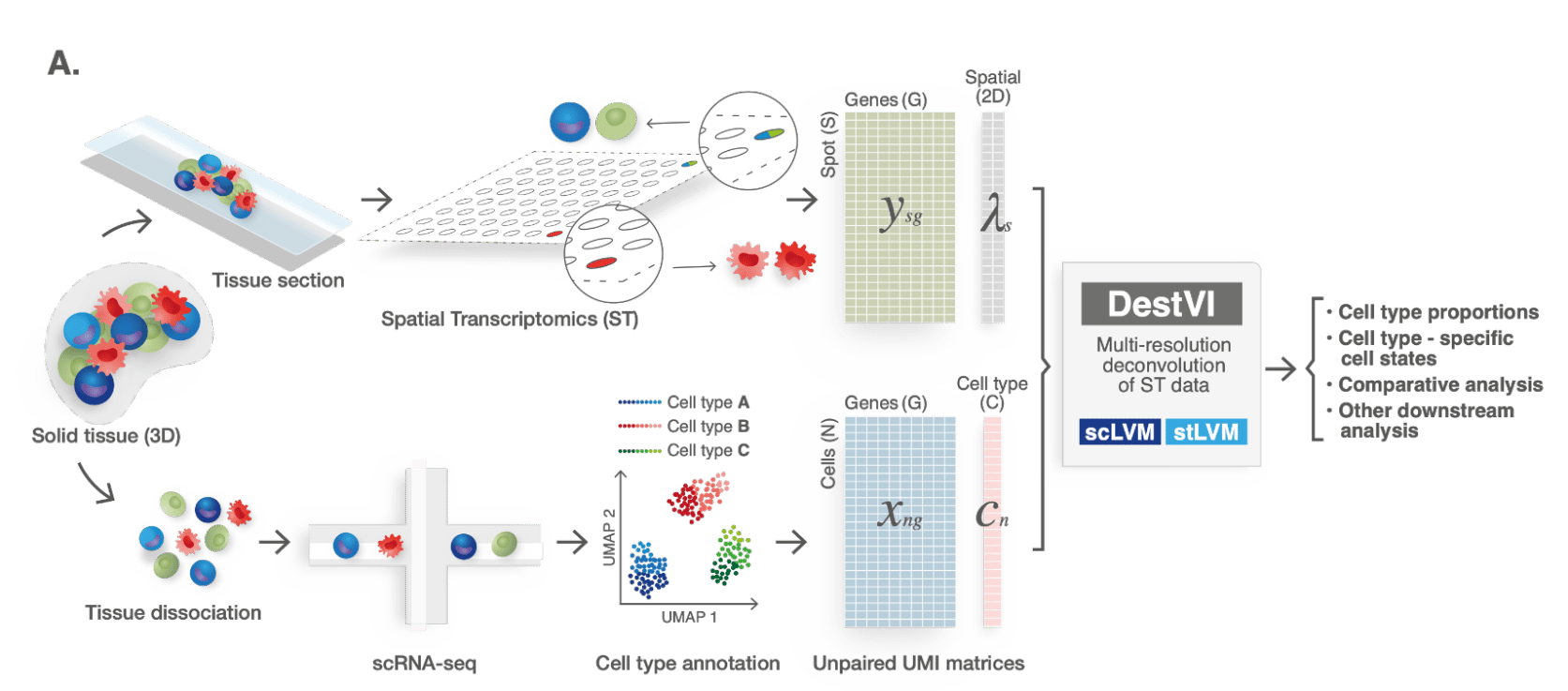
Figure 2: Schematic representation of the ST analysis pipeline with DestVI. Classical deconvolution methods have the same input data, but outputs only cell type proportions and not cell type specific cell states.
We then started developing a computational model for learning continuous cellular states from a scRNA-seq dataset, with the ability to project it onto a spatial transcriptomic dataset (taking into account that each spot may have cells from heterogenous types). The model (named DestVI) builds upon our previous work on scRNA-seq data modeling, and previous deconvolution models (e.g., Stereoscope). Consequently, the model accounts for several types of technical noise, such as library size and overdispersion in the scRNA-seq data, library size and a missing cell type for the ST data, as well as capture efficiency differences between the two assays (refer to Figure 3). DestVI’s inference method scales to large datasets. Additionally, we propose an automated pipeline that seeks, in every cell type, for spatially varying states and a cell-type specific differential expression framework. Both aspects of the pipeline were fundamental in analyzing the new datasets. DestVI is publicly available as part of the scvi-tools package.
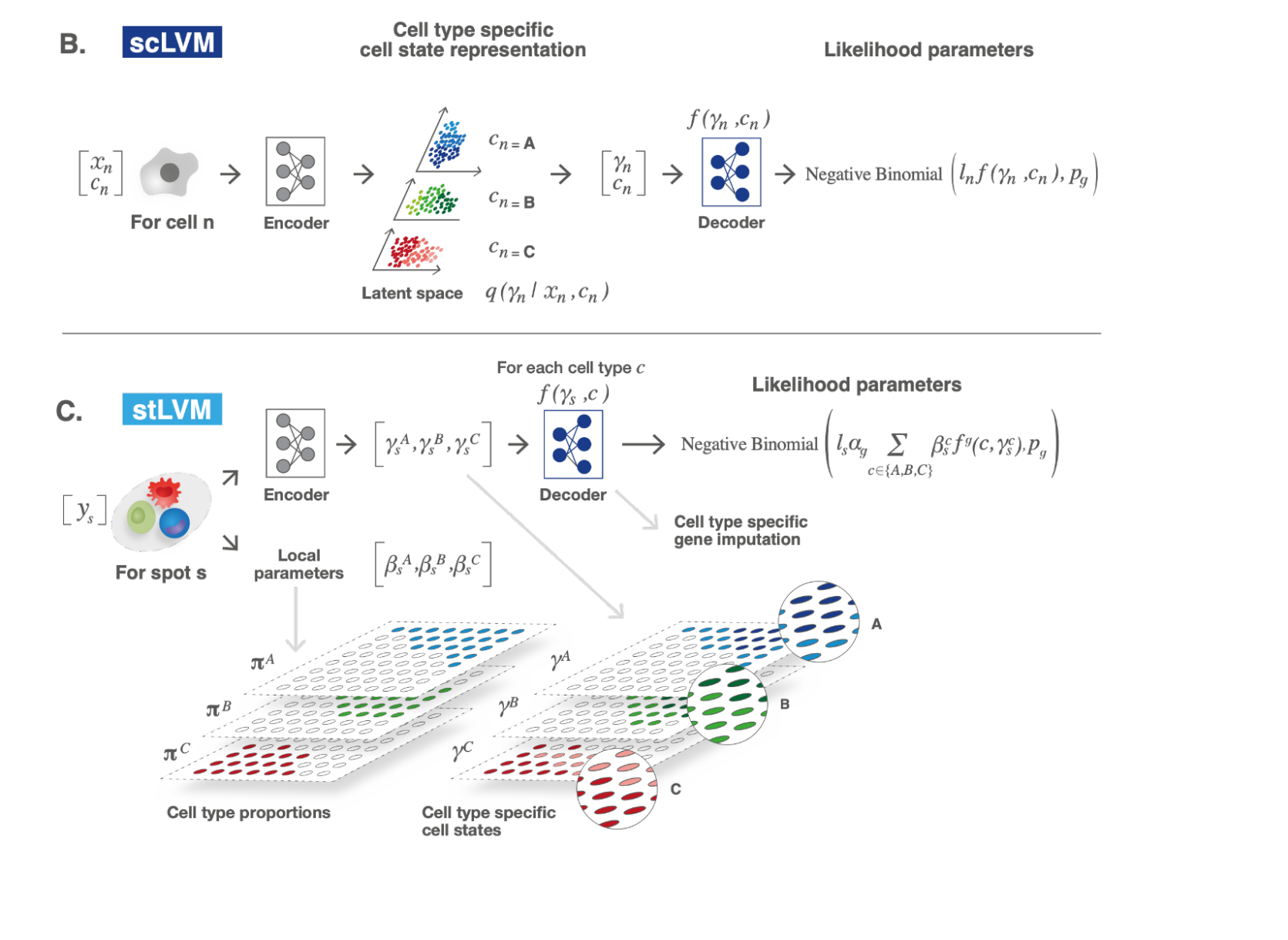
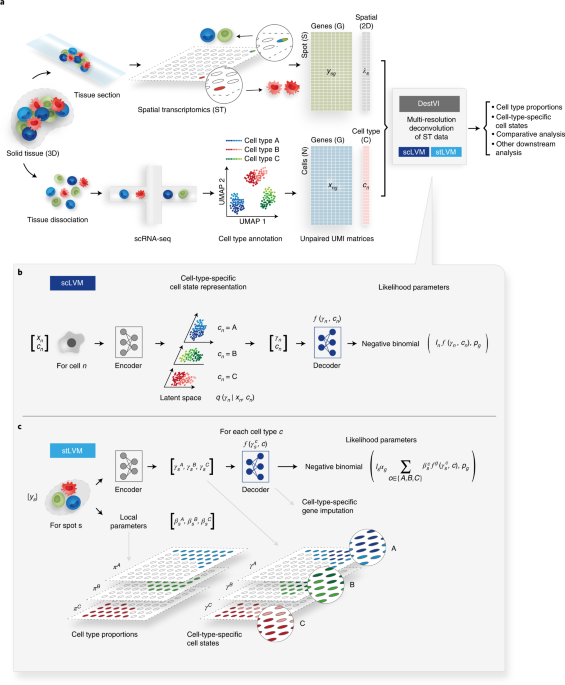
Figure 3: The computational models used in this method. (B) Schematic of the scLVM. RNA counts and cell type information from the single cell RNA-sequencing data are jointly transformed by an encoder neural network into the parameters of the approximate posterior of γ, a low-dimensional representation of cell-type-specific cell state. Next, a decoder neural network maps samples from the approximate posterior of γ along with the cell type information c to the parameters of a count distribution for every gene. (C) Schematic of the stLVM. RNA counts from the ST data are transformed by an encoder neural network into the parameters of the cell-type-specific embeddings γ. Free parameters β encode the abundance of every cell type in every spot. The decoder from the scLVM model maps cell-type-specific embeddings γ to estimates of cell-type-specific gene expression. After training, the decoder may be used to perform cell-type-specific imputation of gene expression across all spots.
Applying DestVI to the new datasets, we were able to localize previously characterized cell states. In contrast with previous methods, we successfully localized the interferon signal of the monocytes to the interfollicular area of the murine lymph nodes, exclusively following exposure to the mycobacteria. On the tumor sample, we spatially mapped a hypoxic phenotype of tumor-associated macrophages to a specific niche of the tumor, that corresponds to the pre-necrotic area. All of those findings were supported by immunofluorescence staining.
The applications of DestVI described in the manuscript focus on the 10x Visium protocol. This commercial assay currently has a low spatial resolution, compared to emerging technologies such as Slide-Seq v2, HDST. Notably, some recent protocols (e.g., Seq-Scope) may even resolve spatial gene expression at the subcellular level. Even in this setting, we anticipate that analysis of data generated from most of those high-resolution methods will still require algorithmic developments such as DestVI, because one cannot always guarantee perfect spatial delineation of single cells (segmentation, registration). We are all extremely excited about technological developments in the spatial transcriptomics field, the release of large spatial data sets (such as the Human Heart Cell Atlas), as well as the scientific discoveries to come.
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in