Building an AI Compass to Navigate Nature’s Hidden Chemistry
Published in Chemistry, Protocols & Methods, and Cell & Molecular Biology

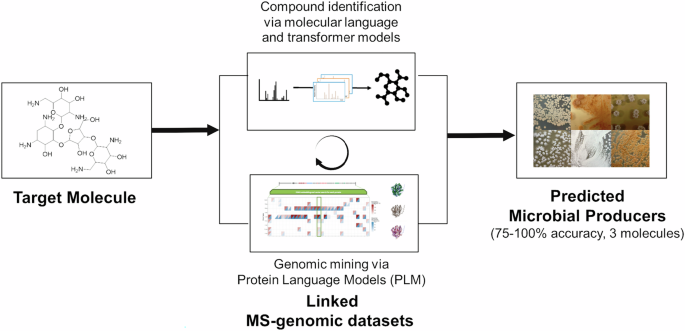
When looking for new bio-based specialty chemicals, the challenge is rarely a lack of samples. The harder question is deciding which strains, extracts or molecular features deserve expensive downstream validation. This is where artificial intelligence can be most useful: not as a replacement for experiments, but as a way to focus them.

The problem: too much data, not enough interpretation
Microbial natural products remain one of the richest sources of biologically active chemistry, but modern discovery campaigns face a paradox. Genome sequencing and mass spectral profiling now generate data at remarkable scale, yet the connection between genetic potential and observed chemistry is still difficult to establish.
On the chemistry side, many mass spectral features cannot be confidently assigned because they do not match existing spectral reference libraries. On the genomics side, many biosynthetic pathways are difficult to decipher because real-world microbial genomes are often incomplete, fragmented, messy, or divergent from known reference clusters.
This creates a practical bottleneck: we may have the data needed to find new chemistry, but not the tools to confidently decide where to look first.
The idea: combine two imperfect but complementary signals
We decided to stop relying on old "rule-books" and instead used language-based AI models to act as translators. Our framework uses two powerful "inference layers":
- The Genomics Layer (The Blueprint Reader): Instead of looking for exact matches in DNA, we used Protein Language Models (PLMs). Think of this like a search engine that understands the meaning of a sentence even if you use different words. These models can recognize the "vibe" of a protein’s function even in messy or incomplete DNA data.
- The WISE Layer (The Chemical Detective): We developed a workflow called WISE (Workflow for Intelligent Structural Elucidation). This uses "generative" AI—similar to the tech behind ChatGPT—to dream up what a molecule might look like based on the shards of information we get from chemical testing. This allows us to identify compounds that aren't in any existing database.

The "handshake": why two are better than one
The real magic happens when these two layers talk to each other. A microbe might have the potential to make a drug (genomics) but might not actually be "turned on" to produce it (metabolomics). By looking for a matching signal—where the blueprint and the product both show up in the same strain—we can prioritize the most promising candidates and ignore the "noise". A key turning point came from our case study on Neomycin B, an antibiotic natural product. Our approach identified four potential microbial producers, and three of them were confirmed to be correct—a 75% precision rate. Interestingly, one of the strains we found had such messy DNA that traditional, rule-based tools completely failed to find the antibiotic gene cluster. Our AI translators, however, were able to "see" the proteins anyway and correctly predicted that this strain was a producer.
What this means for the future
The broader implication is that combining genomic potential with chemical phenotype can reduce the experimental search space and help researchers decide which candidates deserve deeper validation. By decoupling discovery from old libraries and perfect DNA maps, we’ve created a faster, more scalable way to navigate the "dark matter" of microbial chemistry. This technology allows us to search through thousands of strains to find the next generation of medicines, even when the data is sparse or incomplete. We believe this AI-enabled approach will significantly accelerate the discovery of new antibiotics and help us stay one step ahead in the fight against drug-resistant bacteria.
For sectors like specialty chemicals, biotech, agritech, and consumer care, this method also opens up several possibilities:
-
Process Derisking: Accelerating the discovery of alternative host strains or novel enzymes without extensive manual screening.
-
Predictive Modeling: Matching molecular structures to real-world performance to accurately predict and optimize sustainable bio-manufacturing of specialty chemicals.
-
Sustainability Insights: Unlocking natural product diversity beyond existing reference knowledge to replace petrochemically derived compounds
By moving from rule-based lookup tables to predictive, multi-modal AI models, we are transitioning from looking at what nature has done to predicting what nature can do.
References
Tay, D.W.P., Koh, W., Ang, S.J. et al. Accelerating natural product discovery with linked MS-genomics and language/transformer-based models. npj Antimicrob Resist 4, 31 (2026). https://doi.org/10.1038/s44259-026-00206-7
Follow the Topic
-
npj Antimicrobials and Resistance
This journal considers basic, applied, and clinical research that advances our understanding of all aspects of antimicrobials and antimicrobial resistance.

Related Collections
With Collections, you can get published faster and increase your visibility.
Sex and Gender Differences in Antimicrobial Use and Resistance
Publishing Model: Open Access
Deadline: Sep 21, 2026
Molecular mechanisms of antimicrobial resistance
Publishing Model: Open Access
Deadline: Dec 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in