Building gene regulatory maps from single-cell data graphs
Published in Healthcare & Nursing
Synthesis Lectures on Artificial Intelligence and Machine Learning, Vol. 14,
No. 3 , Pages 1-159.
First developed around 2009, single-cell RNA sequencing has allowed detailed inspection of heterogeneous cell states of healthy and diseased tissues1. We can now measure multiple genomic modalities together to read out the matched gene expression, chromatin accessibility, DNA methylation, or cell surface protein level from the same cell2. These modalities are used individually or in combination to define cell states. In order to make sense of hundreds of thousands of features including genes and chromatin-accessible regions, the cell feature vectors can be embedded in low dimensional spaces (e.g., PCA) where cells with similar vector values are closer to each other. In these embedding spaces, clustering algorithms can be used to group similar cells and to identify differentially enriched features in each cluster. Known enriched features (e.g., marker genes or chromatin accessibility peaks) can be used to annotate clusters and unknown features can be investigated for their function and to define novel cell types (Figure 1).

This standard framework, implemented in several popular software packages3,4, has been successfully used to study health or disease states from single-cell sequencing data. However, when we focus on identifying key gene regulatory circuits that define specific cell states or biological processes, such as differentiation, it is not clear how to best integrate the different single-cell level molecular readouts. For example, even if we have multi-omics data that profile both chromatin accessibility and gene expression, it is still challenging to infer transcription factors that drive a particular cell state and their downstream mechanisms. For this purpose, we introduce a new method to integrate cell states with diverse genomic features from single-cell data in a flexible and scalable manner.
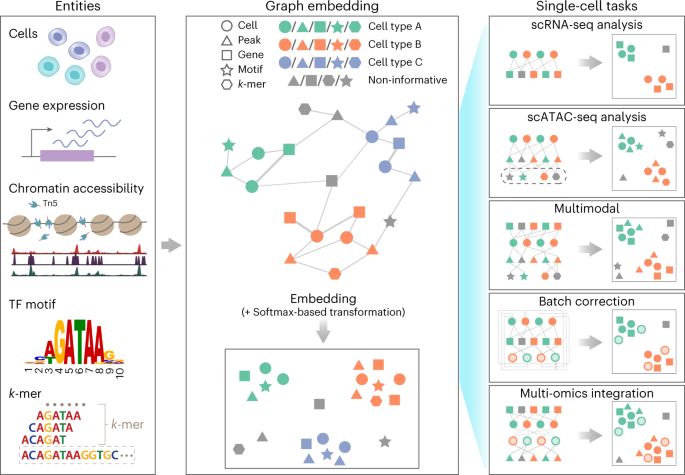
We reason that a graph, a flexible data structure composed of nodes connected by edges, can be the solution to this problem. A single graph with diverse node and edge types, or a heterogeneous graph, can encode complex relationships between different types of nodes. Based on this idea, we developed, SIMBA (SIngle-cell eMBedding Along with features). Our approach constructs single-cell graphs consisting of cells and measured genomic features, such as genes and chromatin-accessible regions (Figure 2). For example, if a gene is expressed in a cell, the gene and cell nodes are linked by an edge with the weight of the gene expression level. The single-cell graph can also have computationally measured or conceptual features as nodes. For example, two chromatin-accessible region nodes can have links to a shared transcription motif node if they both have the transcription motif in their DNA sequences. As such, a heterogeneous graph allows us to encode multimodal single-cell data where the relationship between features is also accounted for.

Similar to existing approaches that can represent a large matrix of cells by features into an embedding space, we can embed nodes of a heterogeneous graph (using a technique called graph representation learning5) into a low-dimensional embedding space such that nodes that share an edge or have similar local neighborhood and structure (e.g., shared neighboring nodes and edges) lie close to each other. In SIMBA, cells and genomic feature nodes are embedded such that adjacent or structurally similar nodes are embedded close by, and the resulting embedding space would have the features/nodes that are specific to the cell subpopulation embedded more closely to the cell subpopulation.
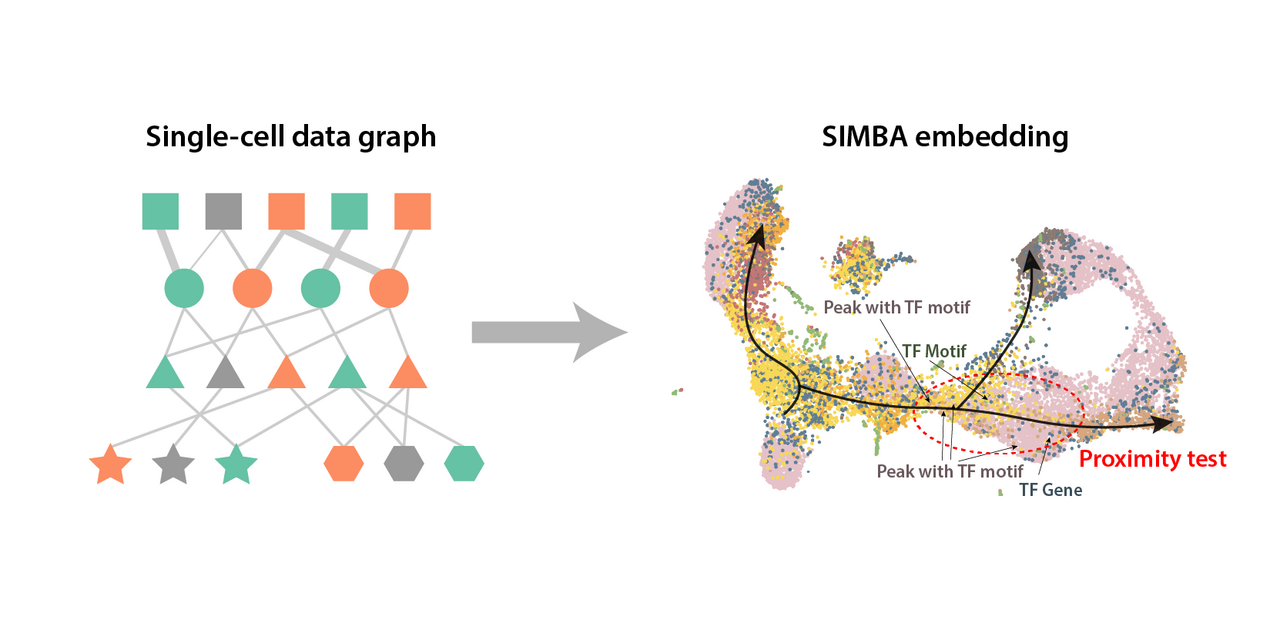
The resulting SIMBA space enables various queries that haven’t been possible without discretizing cell states (Figure 3):
- Which transcription motifs show the highest cell state specificity and in which cell states?
- What are the master regulator transcription factors that show specific expression and specific motif accessibility in the same cell state?
- What are the likely gene regulatory circuits (transcription factor, cis-regulatory element, and target genes) that define a biological process in the single-cell data?
As the embeddings produced by SIMBA are organized spatially according to the similarity of their features, we can answer these questions with a simple proximity search of the embedding space.

We apply SIMBA to a mouse skin SHARE-seq dataset to identify a novel cell state corresponding to a putative master regulator (e.g., the transcription factor RELB and its target genes from mouse skin SHARE-seq data6) (Figure 4). Our results show the power of flexible heterogeneous single-cell graphs and how the graph representation learning approach allows an integrative view of genomic features and cell states that are directly or indirectly related. On top of the novel analyses made possible by graph representation learning, SIMBA shows competitive performances in routine single-cell analysis tasks, including batch correction and modality integration. Besides the evaluation of SIMBA on scRNA-seq, scATAC-seq, and multi-omics datasets, our framework can be readily expanded to incorporate other modalities. The remaining challenges include the integration of bulk data that have higher resolution feature-feature relationship, multi-sample integration, and integration with spatial transcriptomics. Nevertheless, in the era of fast-developing experimental techniques and ample but fragmented data sources, data integration through graph structure, especially in conjunction with single-cell data, holds great potential in studying biological systems in a more holistic view.
 cell type within TAC cells. Transcription motif accessibility and gene expression along with putative target genes’ expression levels are shown on the UMAP.")
SIMBA is implemented as a Scanpy-compatible, user-friendly Python package with comprehensive documentation (https://simba-bio.readthedocs.io/en/latest/). As we use the scalable and efficient PyTorch-BigGraph7 framework, SIMBA is scalable to a scRNA-seq graph with 1 million cells on a laptop.
References
- Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
- Ogbeide, S., Giannese, F., Mincarelli, L. & Macaulay, I. C. Into the multiverse: advances in single-cell multiomic profiling. Trends Genet. 38, 831–843 (2022).
- Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
- Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
- William L. Hamilton. Graph Representation Learning. Synthesis Lectures on Artificial Intelligence and Machine Learning 14, 1-159 (2020).
- Ma, S. et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116.e20 (2020).
- Lerer, A. et al. PyTorch-BigGraph: A Large-scale Graph Embedding System. arXiv:1903.12287 [cs, stat] (2019).
Figure 1 is created with BioRender.com
Follow the Topic
-
Nature Methods

This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Jul 28, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in