Building Large Adsorbate Configuration on Catalytic Surface
Published in Chemistry
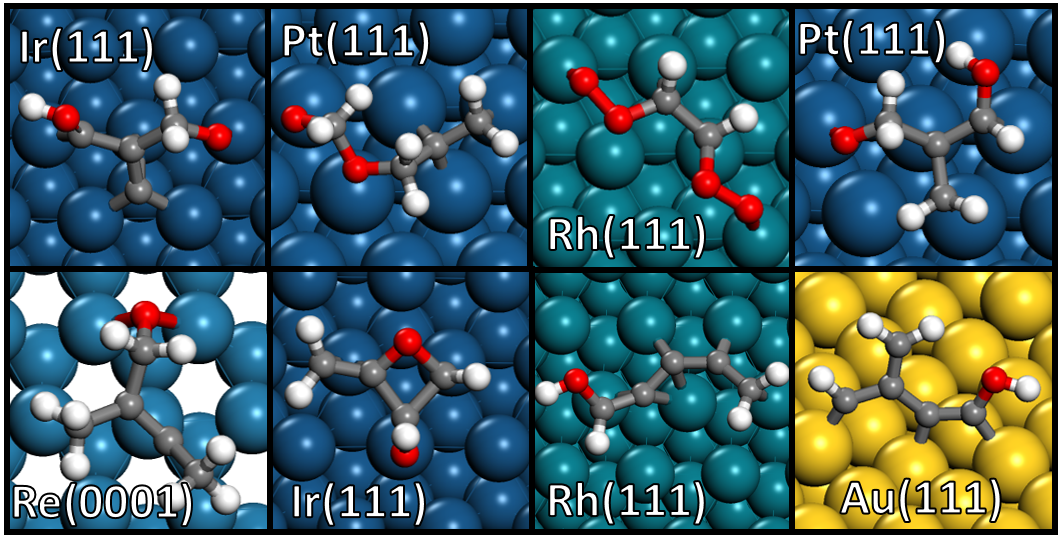

A decade long struggle in studying large molecule conversion
My Ph.D. thesis involved the investigation of lignin monomer conversion on transition metal surfaces. Lignin monomers are derived from biomass that consists of a C6 aromatic ring with various substituents. These molecules are considered large in the world of computational catalysis on metal surfaces due to their large reaction network. On the transition metal surface, elementary reactions such as C-H, C-O, and C-C bond scission and formation occur. By hypothetically applying these reactions over and over to lignin monomers, we can make a reaction network with >~106 reactions and >~105 intermediates (Fig. 1. Chem. Eng. Sci. 2015). This reaction network is too big to investigate using costly quantum mechanical simulations. At the time, Dion was addressing this challenge using the method called group additivity which rapidly predicts molecules’ energy based on each atom’s local environment. I managed to extend this framework to lignin monomers (J. Phys. Chem. C 2016), but the method was far from being practical.
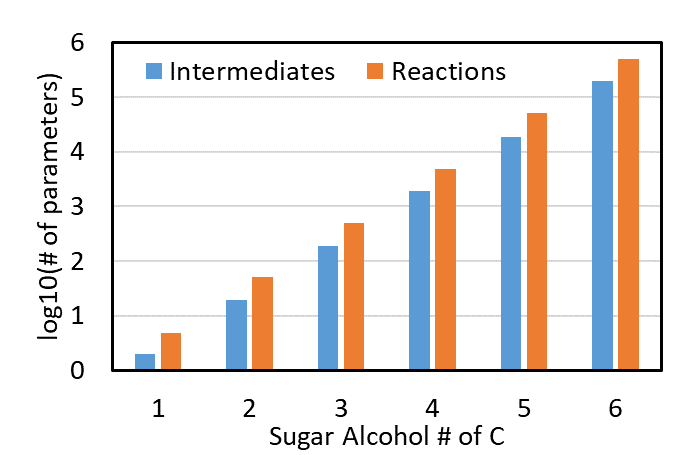
All about adsorption configuration
The frustration and challenges are boiled down to adsorption configurations. An adsorption configuration is an adsorbed structure of a molecule, and, for a large molecule such as lignin monomers, can adsorb with various configurations with different interaction modes due to its aromatic ring (Fig. 2.) The difficulty is that we need to assess them all to find the global ground state, and other local-ground states may be the active state as well (ACS. Catal. 2016). Manually making adsorption configurations for large adsorbates is a tedious, time-consuming task. For the lignin group additivity, I made about 600 data points, which took about 2 full days of repetitive boring work. We also found that to make a group additivity model with decent accuracy, the model needs to know about the adsorption configuration information as well, which means that to use this model, adsorption configurations have to be manually enumerated, making the model inaccessible. Even with all the struggles above, the accuracy was about 3 kcal/mol, which was better than nothing but was not enough to make conclusive results.
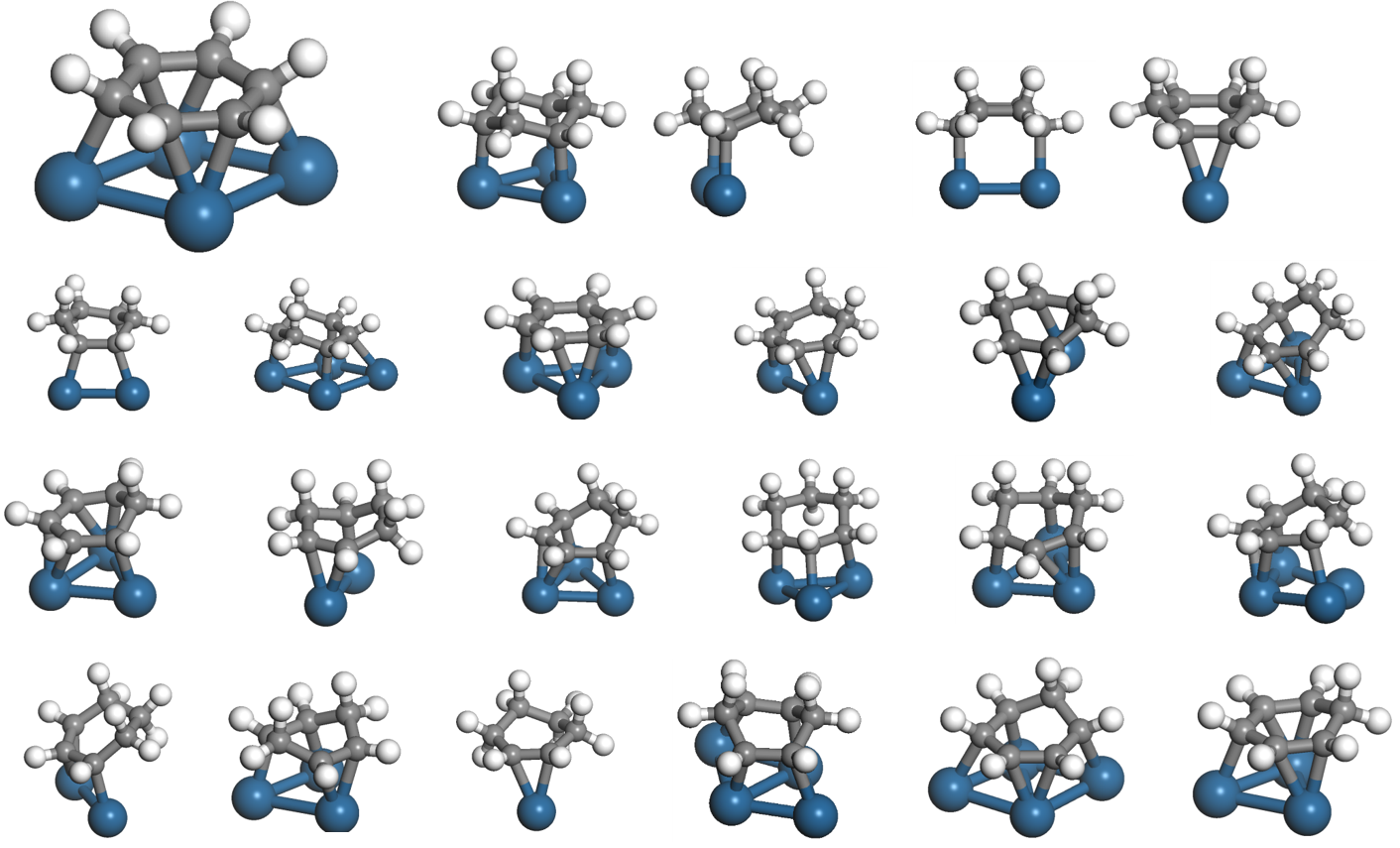
Resolve
To address these challenges, I tried to make a Matlab script that automatically makes configurations, but without much success during my Ph.D. candidacy. The implementation of the molecular graph theory was an important key that was not accessible at the time. I was writing the graph theory code from the scratch in Matlab. Eventually, I had to drop this work, because my time could be better spent working on other projects. Towards the end of my Ph.D. education, the python language and anaconda (website) was gaining popularity, and I found a molecular graph theory package called rdkit (website). With these codes as well as the inspiring organic molecule enumeration work by Ruddigkeit et al (J. Chem. Inf. Model. 2012), I had all the resources to realize the automated configuration generation. It still has taken my whole post-doc to finalize the framework (and the revision to concisely describe the algorithm. Thank you, reviewers!), but this work opens the door to investigating large molecule conversion on transition metal surfaces.
Broader impact
The computational catalysis field has made great advancements. The reaction mechanisms for the electrochemical conversion of various small molecules have been elucidated, and simple activity descriptors have been discovered to screen new catalytic materials (Adv. Mater. 2020). Such investigation for the conversion of the larger molecule has not been possible, application of which include biomass conversion to fuels and chemicals, plastic recycling, Fischer-Tropsch, and CO2 reduction to C3+ products to name a few. This work aims to enable such a powerful application for large molecules by automating the generation of adsorption configurations. This will help us build a large database that will enable data-science methods to help us understand the large molecule catalysis mechanisms and realize the catalyst screening for the large molecule conversion.
The key to the framework
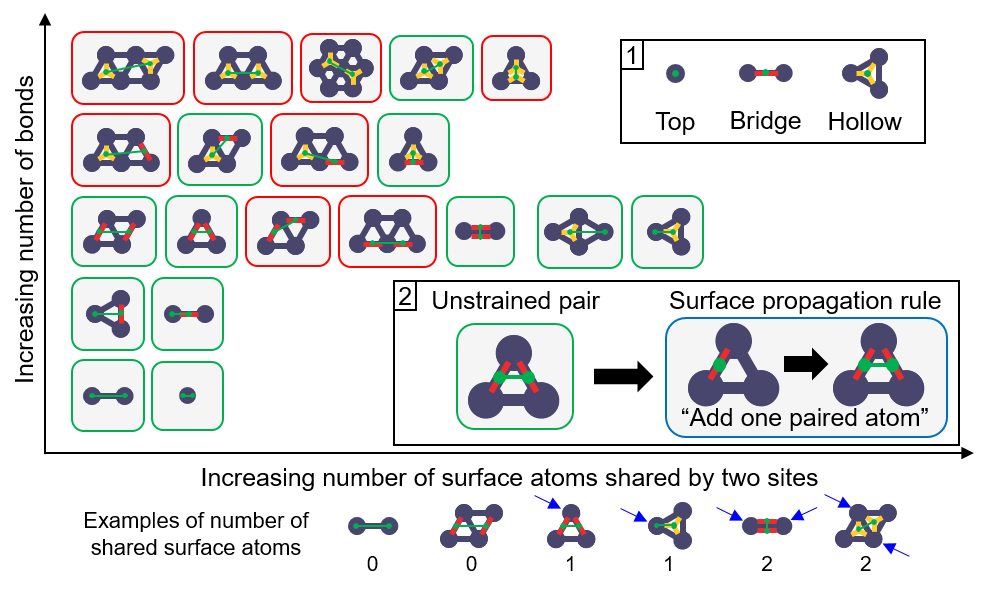
The technical complexity of the framework can distract the readers from the key innovation in the algorithm. We had to make the graph enumeration rules that achieve two goals: (1) the configurations generated using the rules must contain all potentially stable configurations, (2) in doing so, the rules also need to minimize the creation of unstable configurations. To accomplish this, I decided to make rules that add just one atom to the adsorbate and, to keep things simple, the pattern graph only has one adsorbate atom, as opposed to two or more (I tried doing this without much success). From these two simple design choices, the task of making the rules becomes easy. We just need to find all possible two-atom configurations and convert them into pattern and replacement graphs. All two-atom configurations can be found as outlined in the manuscript, where we exhaustively assess two dimensions (number of binding site atoms and the number of shared binding site atoms by two adsorbate atoms) as shown in Fig. 3. With this core of the algorithm, developing the vacuum propagation rules, ring rules, and anchor rules was straightforward.
A pleasant surprise in the stable configurations
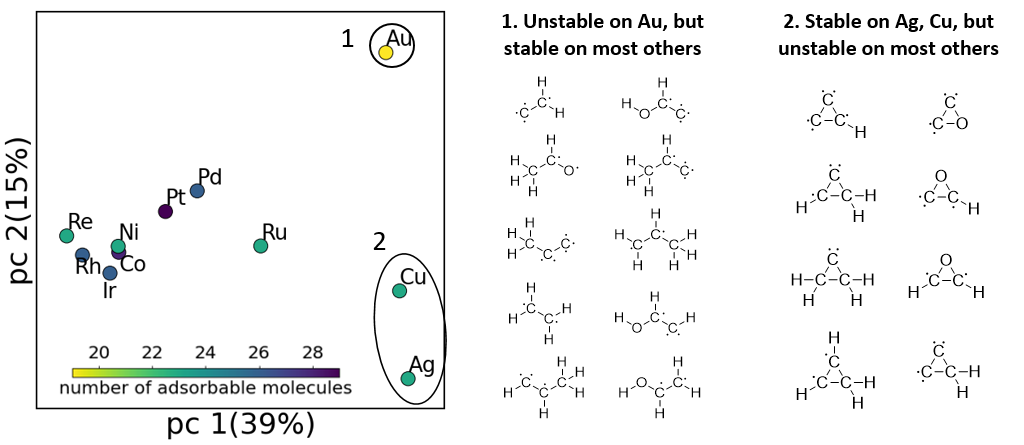
With the newly enumerated data, we performed the principal component analysis of the adsorbate configuration space, which showed that some adsorbates can only adsorb to selected metals. We found that Ag and Cu, which selectively adsorb three atom rings, have good selectivity and activity for ethylene oxide formation (Fig. 4). This indicates a new idea that we can potentially design a highly selective surface catalyst by controlling the stable configurations. To exploit this, we will need to understand how to control the set of stable configurations, which will hopefully be addressed by building a large database using our new algorithm.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in