From manual trek to high-speed journey: Quantifying catalytic performance with CARE
Published in Chemistry, Materials, and Computational Sciences

The design of materials able to accelerate chemical reactions and make industrial processes more sustainable has relied, since the birth of heterogeneous catalysis, on long trial-and-error experimental procedures. The development of the Fe-based ammonia catalyst carried out one century ago by BASF, for instance, involved the manual testing of 3.000 catalyst samples in more than 20.000 experiments, lasting more than 10 years. Although significant advancements in solid-state chemistry, surface science, and screening efficiency have accelerated catalyst design, this task remains a mostly experimental endurance test. In this context, how are atomic-scale simulations positioned? What has been their contribution so far?
Computational scientists simulate reactions to gather insights on the mechanisms behind experimental evidence and characterize them mainly energetically. To understand what simulating catalytic processes on materials means, imagine the world of atoms as a mountain range where the altitude represents the energy. Simulating a surface reaction A → B + C such as a bond-breaking step can be seen as proposing a mountain hike. The first step involves locating the start and final point of the trek, finding how molecules A, B, and C interact with the catalytic surface (the valleys). Evaluating the activation energy of the reaction means hiking from the start (A) to the end valley (B+C) through a mountain pass (the transition state), the highest point of the trek. The thing is that when we start these simulations, we don't start at a trailhead or a parking lot, but it’s as if we were blindfolded and dropped by a helicopter (our chemical intuition) at a random spot on a slope. Atomic-scale simulations help us walking downhill until we find a stable valley, and only then we begin the hike toward the next one. Each reaction in the network represents a single hike, and the better the intuition, the simpler the hike.

The main bottleneck of these tasks is the tedious, manual craft and monitoring required for hundreds of long-running simulations. As the gap in size between reactants and products grows, the number of alternative paths expands exponentially, and to maintain human-feasible timelines, density functional theory (DFT)-based investigations in heterogeneous catalysis have so far been restricted to the activation of small molecules (e.g., CO2, CH4, NH3) in a descriptive setting, where the most common scenario is the one where experimental scientists reach out to the computational counterpart to get help explaining their observations. "Why does this catalyst produce C and not D?" This question is typically answered by developing a reaction mechanism represented by an energy profile, mostly interpreted in qualitative terms. This approach limits simulations to a validating role, as these become biased to the products experimentally observed. The ideal scenario would consider simulations as the starting point of any investigation in order to answer the question: "Given some reactants, a catalyst, and specified operating conditions, what products should I expect?"

Atomic-scale simulations can unveil the phenomena governing catalysis, providing a way to predict the experimental rates and products associated to a specific material before conducting the experiment. If this scenario is still unusual, the main causes are the large amounts of time, resources, and human-demanding tasks associated to DFT. As most of the research time is allocated to propose, prepare, run, and monitor simulations, not enough time is left for "post-processing" their results in a way enabling the extraction of macroscopic observables experimentally accessible, mainly the reaction rate.
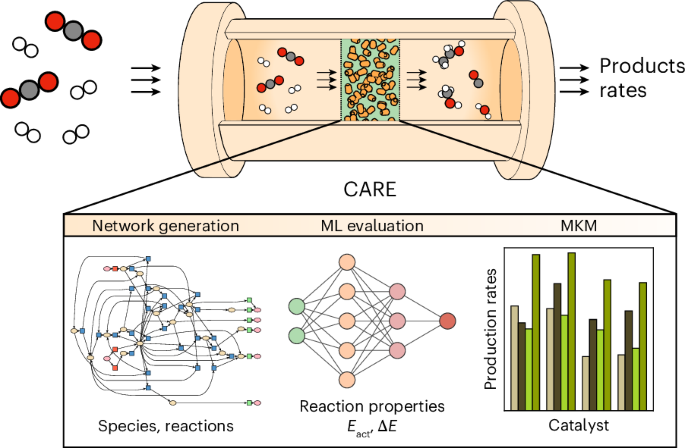
How can we upgrade atomic-level simulations to a guiding role in heterogeneous catalysis? Replacing DFT with faster machine learning (ML) predictive models is an option. The advent of ML created a fascinating alternative not based on the direct study of physical-chemical phenomena, but on a purely data-driven fashion. In the same way a catalyst speeds up a reaction, ML must accelerate catalyst design. The work published by Morandi, Loveday et al. in Nature Chemical Engineering is a getaway towards this direction. The authors present the Catalytic Automated Reaction Evaluator, CARE, an open-source software for the automated generation and evaluation of reaction networks in heterogeneous catalysis. CARE provides the toolbox required to develop the methodologies needed for predicting macroscopic reaction rates starting from atomic-scale simulations.
Think of CARE as a special vehicle built around a high-speed ML engine. While this engine releases the power to navigate thousands of "atomic roads" in seconds, CARE provides the steering wheel and GPS to ensure the power unit does not drive off a cliff. By automating the search for "valleys" (intermediates) and "passes" (reactions), it transforms a manual, error-prone trek into an automated, high-speed journey, allowing researchers to focus on the destination—the quantification of catalytic performance—rather than the difficulty of the hike.

What can you do with CARE
CARE allows to automatically generate and evaluate reaction networks involving C, H, and O containing species. As for the catalyst, the framework is not constrained to a specific class of materials. The material domain that can be investigated with CARE only depends on whether the ML evaluator of choice can process it. For instance, GAME-Net-UQ targets transition metal catalysts, while popular ML Interatomic Potentials (MLIPs) like MACE-MP-0 and EquiformerV2 target most elements in the periodic table. CARE enables to generate networks not only in thermal conditions, but also in electrochemical conditions.
CARE includes a module for running kinetic simulations over the generated networks, predicting the evolution in time of the participating species and therefore the catalytic performance of the material under study.
How large can the networks generated with CARE be?
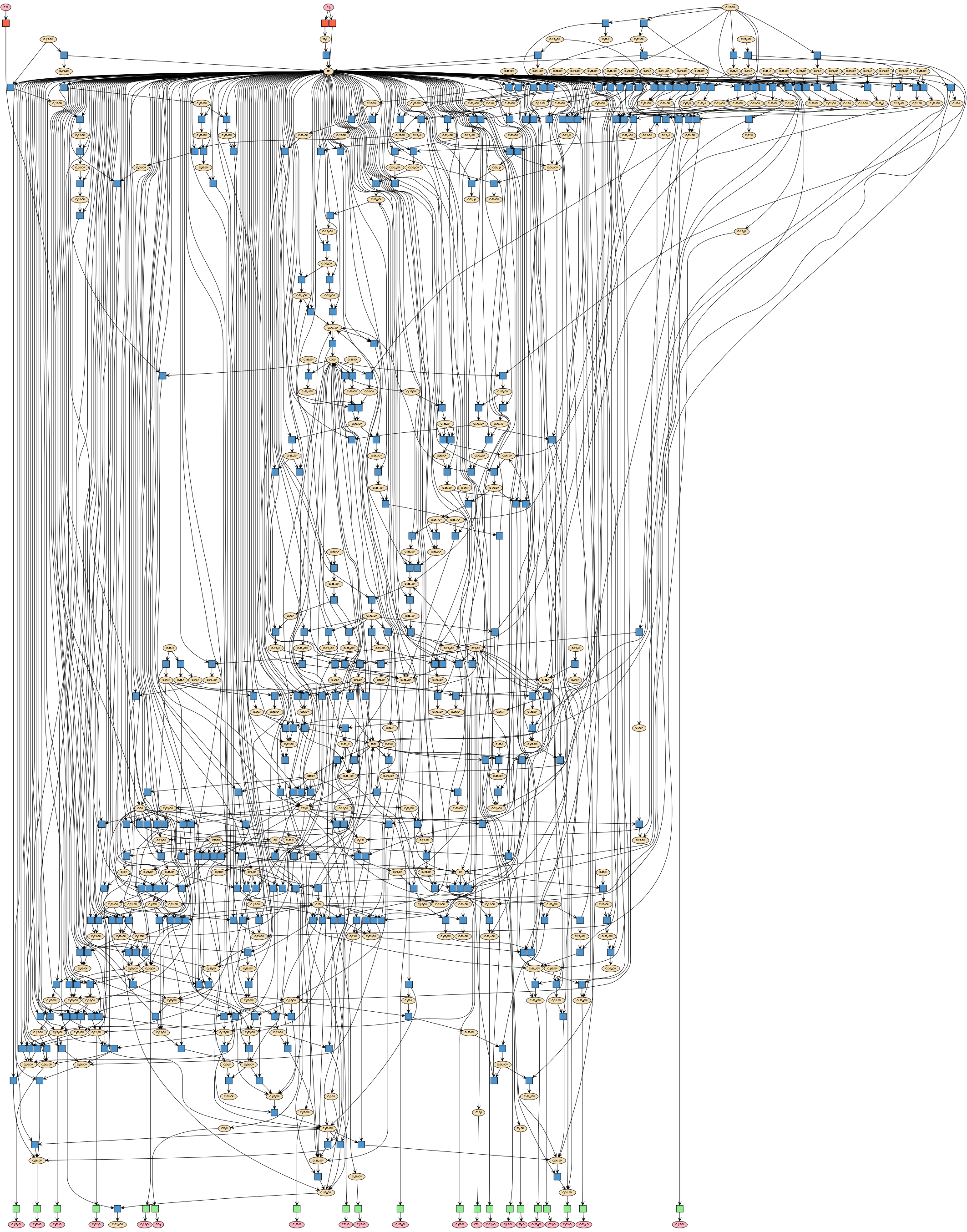
Very large. We achieved to generate in hours a network representative of the Fischer-Tropsch reaction -- producing synthetic fuels starting from carbon monoxide and hydrogen-- including 370.000 elementary steps, without relying on supercomputers. Additionally, we show how we achieved to run microkinetic simulations on sub-networks of 37.000 reactions up to steady state conditions and obtain trends in agreement with experiments. For you to know, the network displayed below contains around 250 reactions, 0.5% of the simulated networks.
In summary, end-to-end ML-powered frameworks such as CARE can catalyze the development of new methodologies bridging the existing gap between theory and experiments, elevating simulations in catalysis to a steering role. We envision a future where digital twins of heterogeneous catalysts are employed for real-time predictions in high-throughput experimentation, accelerating the time-to-market for more sustainable catalysts.
Follow the Topic
-
Nature Chemical Engineering
This is a new monthly online journal dedicated to publishing the most significant original research, commentary and analysis of direct relevance to the diverse community of chemical engineers.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
great work!