ChromID identifies the protein interactome at chromatin marks
Published in Bioengineering & Biotechnology

Large eukaryotic genomes are tightly packaged to fit into the tiny space of a nucleus. To achieve this, DNA is wrapped around unique proteins called histones, resulting in a DNA-protein complex called chromatin. Chromatin is enzymatically modified by the addition of a plethora of chemical modifications, including acetyl, methyl, or phosphate groups, some of which are known to play essential roles in replication, transcription, chromatin compaction, and DNA damage repair. Hundreds of specialized proteins are predicted to bind or interact with these modifications directly, influencing the protein makeup of the chromatin of each cell type of our body. Importantly, absence and wrong placement of these modifications are associated with numerous diseases. Therefore, understanding how these chemical modifications influence the organization and protein composition of chromatin has become a central focus of biomedical research (1).
Two current challenges are i) to identify which proteins associate with the genome based on the local chromatin modifications, and ii) to dissect the mechanisms of how reader proteins bind to the genome.
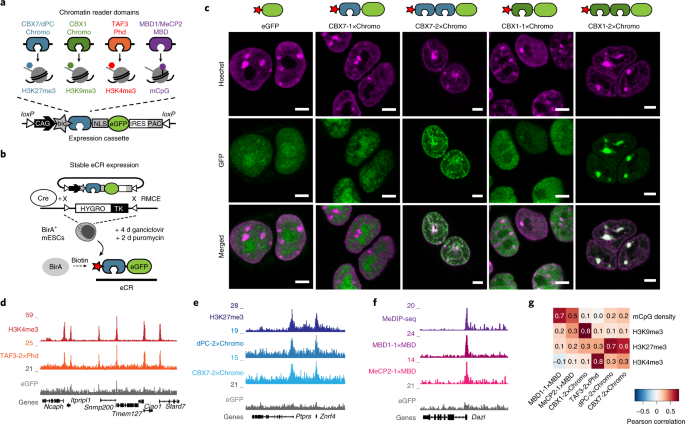
In our recent article in Nature Biotechnology, we present new methods to address both challenges at unprecedented depth and in a highly reproducible manner. To tackle these questions, we used parts of proteins, so-called “reader domains”, that interact with specific chromatin modifications or DNA sequences. These reader domains are present in many regulatory proteins and are required for their site-specific localization to the genome. However, for the majority of these domains, the target histone modifications are unknown. To address this problem, we wanted to develop a simple method that enables us to systematically investigate the binding preferences of isolated reader domains in living cells. As a proof of concept, we first used natural reader domains of well-studied chromatin-interacting proteins as modular building blocks to develop so-called “engineered chromatin readers” (eCRs). By stably expressing eCRs in mouse embryonic stem cells and measuring their subnuclear localization, genomic distribution, and histone–modification–binding preference, we could demonstrate their selectivity for chromatin modifications and the suitability of eCRs to study chromatin readout in living cells. Notably, we found a simple design principle (multivalency) to yield selective histone-mark readers, which has also been shown by other studies (2-4). The modular architecture of eCRs allows us to study virtually any reader domain found in nature, and build eCRs that are selective for combinatorial marks. We are excited to use eCRs soon to catalog the targets of many unknown reader domains and understand their role in chromatin regulation.
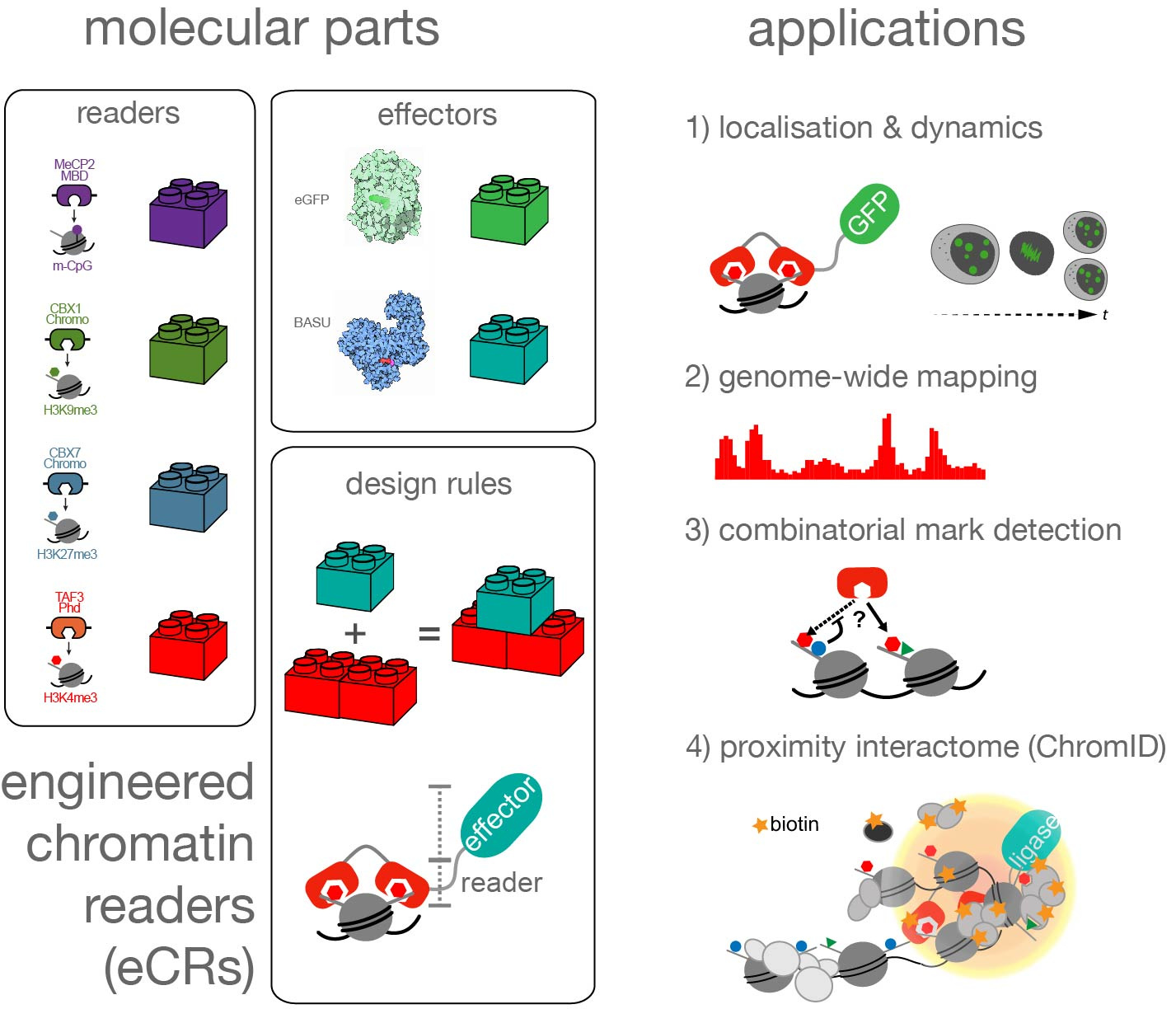
Lastly, to identify which proteins associate with certain chromatin marks, we used eCRs to recruit a bacterial “biotin labeling” enzyme to distinct regions in the genome based on chromatin readout. As a result, the proteins associated with or bound near the respective target chromatin modification were labeled with biotin. This strategy allowed us to isolate biotin-labeled proteins and detect them via mass spectrometry. By applying this method, which we termed ChromID, we could identify a variety of proteins associated with several histone and DNA modifications in mouse stem cells. Our results revealed previously unknown crosstalk between specific chromatin environments, lead to the discovery of novel proteins associated with individual chromatin modifications, and uncovered the proximal interactome at bivalent promoters marked by both H3K4me3 and H3K27me3. The applicability of ChromID in living cells, as well as eCRs as synthetic readers, further opens the exciting possibility to perform similar experiments in living animals to chart the epi-proteome during dynamic cellular processes and development.
In summary, identifying proteins bound at distinct chromatin regions decorated with specific modifications has been a long-standing challenge (5). With ChromID, we demonstrate the potential of coupling selective chromatin readers with proximity biotinylation to overcome this challenge. Finally, our study provides the chromatin community with a set of chromatin-mark interactomes and tools to track essential histone modifications and DNA methylation in living cells.
The full paper can be accessed through the link below:
https://www.nature.com/articles/s41587-020-0434-2
The Baubec lab website: http://www.baubeclab.org
References:
1. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489-99.
2. Mauser R, Kungulovski G, Keup C, Reinhardt R, Jeltsch A. Application of dual reading domains as novel reagents in chromatin biology reveals a new H3K9me3 and H3K36me2/3 bivalent chromatin state. Epigenetics Chromatin. 2017;10(1):45.
3. Delachat AM, Guidotti N, Bachmann AL, Meireles-Filho ACA, Pick H, Lechner CC, et al. Engineered Multivalent Sensors to Detect Coexisting Histone Modifications in Living Stem Cells. Cell Chem Biol. 2018;25(1):51-6 e6.
4. Tekel SJ, Vargas DA, Song L, LaBaer J, Caplan MR, Haynes KA. Tandem Histone-Binding Domains Enhance the Activity of a Synthetic Chromatin Effector. ACS Synth Biol. 2018;7(3):842-52.
5. Wierer M, Mann M. Proteomics to study DNA-bound and chromatin-associated gene regulatory complexes. Hum Mol Genet. 2016;25(R2):R106-R14.
Cover photo: adapted from David S. Goodsell
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in