Controlling Helicity and Optical Properties in π-Conjugated Backbones by Tuning Aromatic Unit Twist.
Published in Chemistry


In the field of organic electronics, π-conjugated oligomers and polymers commonly replace inorganic semiconductors as the active layer. The main advantage of organic electronic materials is that, in principle, their properties can be tailored by chemical means to obtain materials with small bandgaps (essential for efficient organic solar cells), having different colors (e.g., for use in organic light-emitting diodes and smart windows) and with high solubilities (important for their processability).
Many groups, including our own, have invested years of research in trying to actualize the potential of organic electronic materials as a means of obtaining a wide range of desired properties in a readily controllable manner. One approach that has been the subject of considerable research is side-group engineering, in which electron-accepting or -donating groups, solubilizing groups, or chiral units are attached to a π-conjugated organic backbone (Figure 1a). Alternatively, main-chain engineering is undertaken, with different units combined within the π-conjugated backbone to obtain desired properties. A third approach involves engineering the molecular conformation of the backbone to obtain desired properties. Conformational engineering commonly relies on non-covalent interactions or on functionalization with sterically bulky side-groups to achieve conformational locking and thereby control the degree of rotation around the backbone’s inter-unit single bond (Figure 1b). Such rotation can produce interesting properties, such as increased intersystem crossing to the triplet excited state (which is important for light emitting devices), via processes such as thermally-activated delayed fluorescence (TADF). It can also introduce an intrinsic helical chirality to the π-conjugated backbone, leading to chiral conducting polymers. Such polymers are greatly desired because of their wide range of potential applications, from spin filters to magneto-optic materials. However, the conformational engineering approach is commonly avoided since it drastically reduces π-conjugation in the backbone. Consequently, although helicity dominates the most fundamental molecules of life, such as DNA and proteins, it is less common in the context of π-conjugated polymers. Therefore, one crucial question is how π-conjugation, which requires planarity for good p orbital overlap, can be caused to coexist with helical chirality, which require deviation from planarity.
For many years, our group has sought a way to induce twist into the π-conjugated backbones of organic materials without impairing their conjugation. We considered the key to achieving this may lie in abandoning twisting around the single bond in favor of polymerization of an intrinsically twisted aromatic unit (Figure 1c). Exploring this approach meant we first needed to synthesize suitable monomeric units and then develop a method to oligomerize and polymerize them.
Focusing on acenes, because they are essentially one dimensional nanographenes and therefore the most fundamental polyaromatic hydrocarbon, we considered several candidates and investigated them computationally. We then endeavored to find synthetic methods for the most promising units. We showed that acenes can be twisted to a controlled extent by diagonally connecting the ends of the molecule using a covalent alkyl-ether tether of variable length. The degree of twisting induced by the tether directly affects the optical and electronic properties of the resulting twistacenes, with electron paramagnetic resonance (EPR) studies showing that electron delocalization is hardly affected by the twist. In the present work, we addressed the considerable synthetic challenge of extending the intrinsically helical acene unit to introduce oligomers of various length and twist, which we finally achieved through linkage via acetylene end-units, which were coupled to form oligomers of various lengths. The helicity of these oligomers is controlled by the aromatic unit twist, which is in turn controlled by the tether length (Figure 1d).
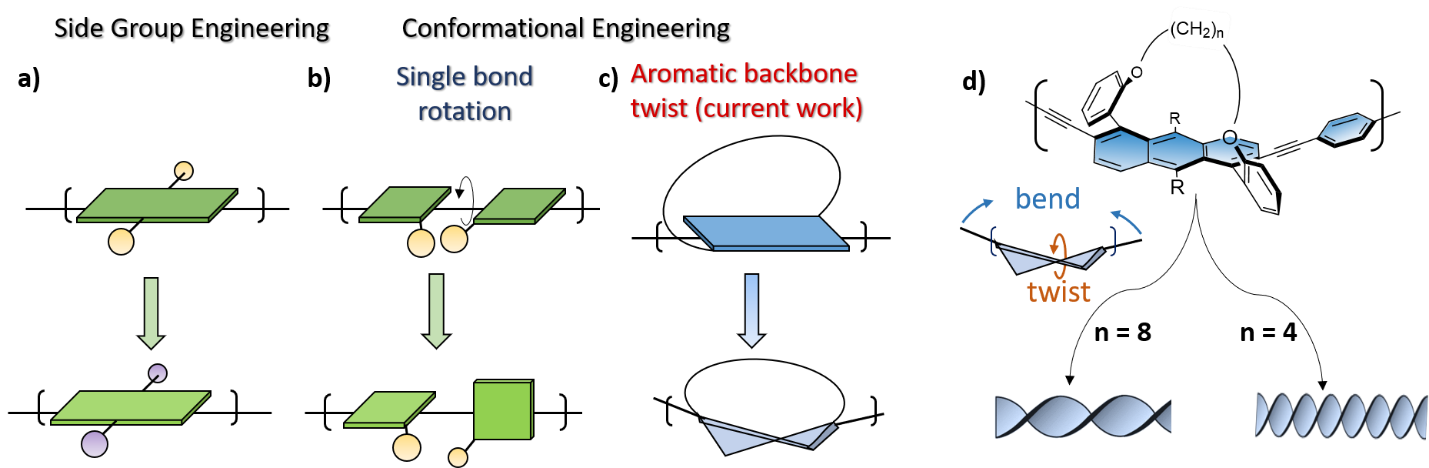
Figure 1. Approaches to tuning the properties of π-conjugated backbones. (a) Side-group engineering. Conformational engineering of the backbone by (b) single bond rotation and (c) aromatic backbone twist, as introduced by our group. (d) Molecular representation of the tethered twistacene π-conjugated oligomers depicted in (c). The length of the tether ((CH2)n, where n = 4, 6 or 8 methylene groups) determines the primary and secondary structures of the twistacenes upon twisting and bending, respectively, and eventually long backbones (polymers) adopt a secondary helical conformation exhibiting varying degrees of helicity. R = CC(CH3)3.
Having introduced π-conjugated backbones with different conjugation lengths and twist angles, we could explore the effect of both factors on the electronic, optical and chiroptical properties of the novel materials. We found that both backbone length and twist affect electronic and optical properties. Synergistic effects of increasing backbone length and twist affect properties such as the optical bandgap and the fluorescence maxima (Figure 2a), whereas antagonistic effects are observed for other properties, such as the extinction coefficient (Figure 2b). For fluorescence quantum efficiency, backbone twist has a greater effect than backbone length (Figure 2c).
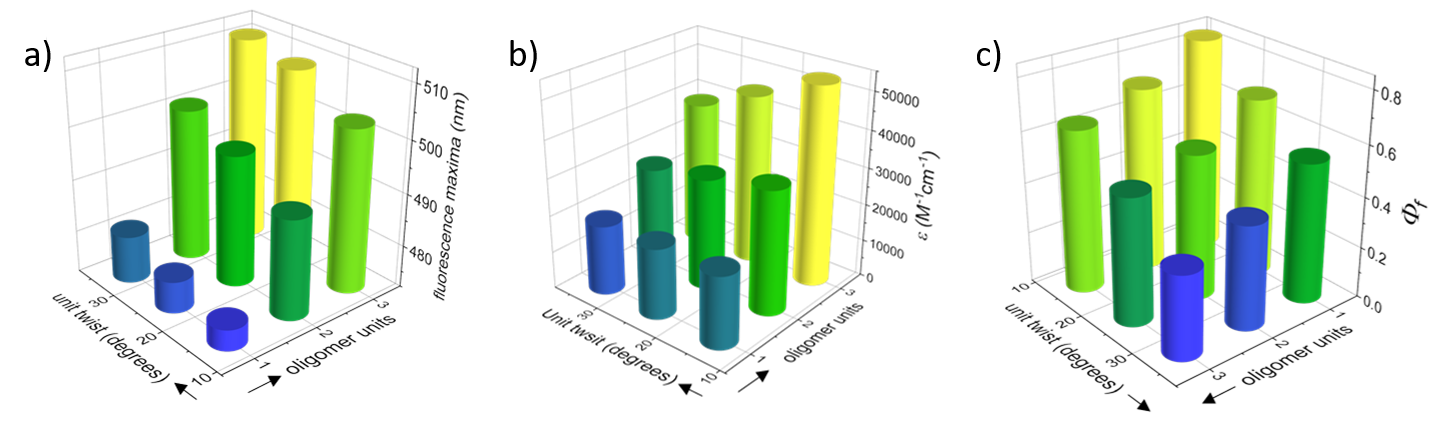
Figure 2. 3D graphs displaying the effect of backbone unit twist (Y axis) and backbone length (X axis) on (a) extinction coefficients at maximal absorption; (b) fluorescence maxima; and (c) fluorescence quantum yields (Φf).
As tethering induces not only backbone twist but also bends the backbone to various degrees, the polymer adopts a helical secondary structure. The secondary helical structure is expressed in chiroptical properties: backbones formed of units with a shorter tether (greater twist) show a non-linear increase in their maximum ellipticity. The non-linear nature of the increase suggests the occurrence of chiral amplification of the effects of the primary and secondary helical structures on incident circularly polarized light, with both helicities (and, consequently, their amplification) controlled by tether length. Indeed, calculated structures show that the tether controls not only the primary structure, but also the degree of helicity of the secondary structure, allowing us to tune helicity while maintaining π-conjugation (Figure 3).

Figure 3. Conceptual representation of how tuning the tether length of the repeat aromatic unit results in oligomer or polymer backbones with different helicities. The two middle structures are calculated nonamer skeletons with end-to-end anthracene twists of either 10° or 40°. The pitch of the secondary helical structure is controlled by the tether length.
Overall, the introduction of extendable twistacene units with controllable twist paves the way to the first synthesis of π-conjugated oligomers and polymers with desirable electronic and optical properties that can be tuned without impairing π-conjugation, and whose backbones possess inherent primary chirality and gain secondary chirality upon extension.
For further details, please refer to our article in Nature Communications: https://www.nature.com/articles/s41467-022-28072-7
For more information of the Gidron research group, please visit: http://chem.ch.huji.ac.il/gidron/
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Mar 24, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in