CRISPR-free, strand-selective DNA base editing by introducing nickase
Published in Bioengineering & Biotechnology

Mitochondria, as the "powerhouse" of the cell, play an important role and are the only organelles in mammalian cells, other than the nucleus, that store genetic information. Mutations in the mitochondrial genome often cause severe diseases, such as Leber hereditary optic neuropathy (LHON)1. There are 97 confirmed mitochondrial genetic diseases, of which 93 are caused by point mutations (MITOMAP). Since electron transfer in mitochondria pumps protons out of the mitochondrial matrix, the matrix has a strong negative charge, which hinders the entry of nucleic acids with the same negative charge2. ZFN and TALEN, whose functional components are proteins, can enter mitochondria and perform gene editing under the guidance of mitochondrial localization signals. In 2018, two groups used ZFN and TALEN to knockdown the mutated genome in mouse mitochondria3,4. However, knockdown cannot treat homozygous mitochondrial mutations, nor can it change the base of the mitochondrial genome.
Base editors, and their derivatives, have shown great potential in the treatment of genetic diseases. Base editing is developed based on the CRISPR-Cas system, which is because the deaminases used in base editors are ssDNA deaminases. With the help of Cas9 protein and sgRNA, one strand of the target DNA double helix is exposed and used as a substrate for deaminase, enabling effective base conversion5. In contrast to the CRISPR-Cas system, the Zinc Finger (ZF) and Transcription Activator-Like Effector (TALE) only have the activity of binding to DNA double strands, but do not unwind DNA double helix6,7. Therefore, prior to the widespread adoption of CRISPR, the combination of ssDNA deaminases with ZF or TALE was unable to achieve effective base editing of DNA8.
In 2020, Joseph D. Mougous's and David R. Liu's lab discovered a deaminase, DddA, that can act on double-stranded DNA, whereas most previously known DNA deaminases only act on single-stranded DNA. Based on DddA, they developed a new base editor and applied it to edit the mitochondrial genome9. Furthermore, Kim Jin-soo's lab also achieved mitochondrial genome editing using DddA and TadA8e10. Recently, many base editors based on DddA, and its homologs and variants have been developed11,12. Since the deamination reaction requires the base to flip out to the active pocket of the deaminase, this process breaks the base pairing of DNA. Therefore, we speculate that DddA may have the ability to unwind the DNA double helix and deaminate the exposed bases. Since DddA does not distinguish between the two DNA strands, base editors based on DddA edit both strands of DNA, which differs from CRISPR-based base editors that only act on one strand of DNA. In addition, DddA also interacts with CTCF in the nucleus, thus off-target editing in the nucleus may occur13.
DddA is only capable of achieving C to T base editing and does not distinguish between the two DNA strands. Therefore, we started attempting A to G base editing on mitochondrial DNA in 2021. We tried various methods, including using a Cas12-based base editor with very short sgRNA, mutating DddA to enable it to deaminate adenine, engineering TadA protein to make it capable of acting on double-stranded DNA, searching for DddA homologous proteins with A to G ability, among others. Unfortunately, none of these attempts yielded positive results. Since the substrates of deaminases from A to G currently developed are all single-stranded DNA, we speculated that the formation of a nick on DNA might generate transient ssDNA, which could serve as a substrate for ssDNA deaminase, so we focused on using a nickase.
 Fig.1 | CRISPR-free, strand-selective DNA base editing by introducing nickase. a,b, Mitochondrial A-to-G editing efficiency treated with paired TALE-TadA8e(V106W) (a) and Left-TALE-MutH and Right-TALE-TadA8e(V106W) (b) at MT-RNR2 site. c, A speculative working model for mitoBEs. TALE-nickase binds and nicks the dsDNA. The nicked dsDNA is prone to form ssDNA, which allows the TALE-deaminase to deaminate. Following DNA repair and replication, editing results on the non-nicked strand are preserved. TadA8e(V106W) was used for mitoABEs in this model. This model also applies to mitoCBEs.
Fig.1 | CRISPR-free, strand-selective DNA base editing by introducing nickase. a,b, Mitochondrial A-to-G editing efficiency treated with paired TALE-TadA8e(V106W) (a) and Left-TALE-MutH and Right-TALE-TadA8e(V106W) (b) at MT-RNR2 site. c, A speculative working model for mitoBEs. TALE-nickase binds and nicks the dsDNA. The nicked dsDNA is prone to form ssDNA, which allows the TALE-deaminase to deaminate. Following DNA repair and replication, editing results on the non-nicked strand are preserved. TadA8e(V106W) was used for mitoABEs in this model. This model also applies to mitoCBEs.
We designed TALE-Nickase and TALE-Deaminase to target the mitochondrial genome and found that effective mitochondrial DNA base editing can be achieved using this combination. First, we tested nickase MutH. We used TALE-MutH and TALE-TadA8e(V106W) with mitochondrial localization signals to target three sites in the mitochondrial genome and found effective A-to-G editing at all test sites. We named this system the mitochondrial DNA base editor (mitoBE). Further investigation revealed that the editing induced by TALE-MutH and TALE-TadA8e(V106W) is strand-selective, and the editing results tend to be retained on the DNA strand not cleaved by TALE-MutH (Fig. 1). By controlling the cleaved strand of TALE-MutH, the strand to be edited in the DNA double helix can be controlled, making the editing results of mitoBE more specific compared to those of the DddA-based base editor.
MutH has its own sequence preference, and it cleaves one of the 5’-GATC-3’ palindrome sequences. To expand the editing scope of mitoBE, we mutated MutH based on structural information and expanded its editing range to 5’-GATN-3’. We named this mutant MutH*. Compared to MutH, MutH* cleavage sites occur twice every 40 bp in the mitochondrial genome, greatly expanding the editable range of mitoBE. In addition, through the screening, we found that the C-terminal of Nt.BspD6I and FokI-FokI(D450A) are recognition-sequence-free nickases that can be applied to the mitoBE system (Fig. 2).
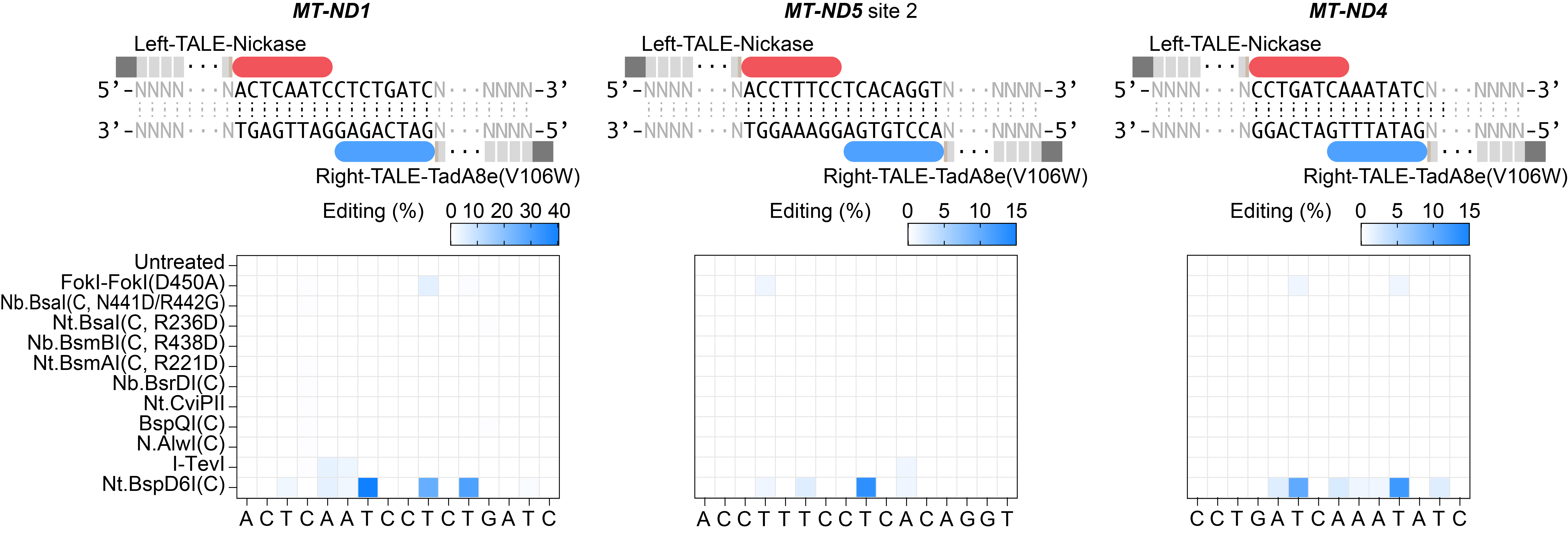
Fig.2 | Screening nickases without recognition sequence restriction for mitoBEs. Nickases without sequence restriction or with extensive recognition sequences were screened for mitochondrial base editing at three sites. The mean values from n = 3 biologically independent replicates are shown.
SsDNA deaminases have been widely used in CRISPR-based base editors, and mitoBEs serve as a toolbox where almost all ssDNA deaminases can be incorporated. For example, we found that TALE-MutH and TALE-rAPOBEC1-2xUGI can efficiently achieve C-to-T base conversion at target sites. Compared with DdCBE, mitoBE has extremely high strand specificity (Fig. 3). Furthermore, through whole-genome sequencing, we found that mitoBE causes minimal off-target editing in both mitochondria and the nucleus, indicating its high specificity.
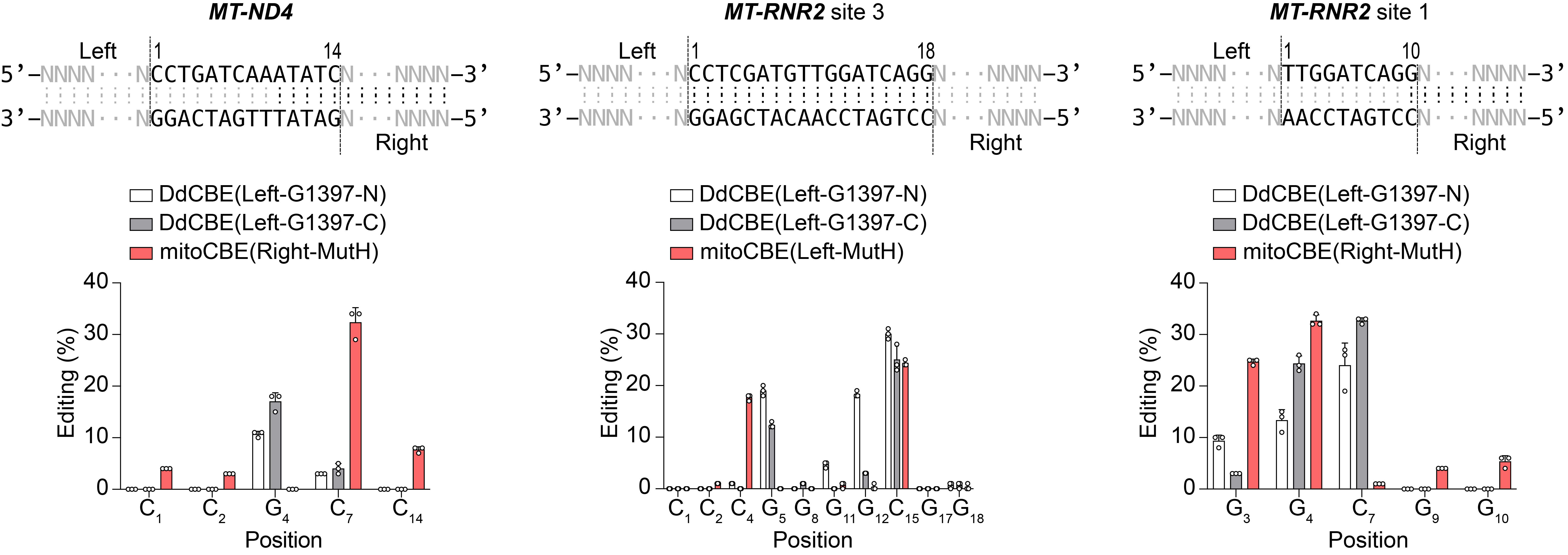
Fig.3 | Compare the editing profiles of mitoCBEs and DdCBEs at MT-ND4 (Left), MT-RNR2 site 3 (Middle), and MT-RNR2 site 1 (Right). Data are presented as mean values ± s.d. of n = 3 independent biological replicates.
LHON is a disease caused by mitochondrial gene mutations, and we attempted to use mitoBE to correct the error DNA base in this disease. First, we tested and found the use of circular RNA-encoded mitoBE to achieve efficient mitochondrial DNA strand-selective base editing. Then we used patients-derived cells and found that the editing efficiency of circular RNA-encoded mitoBE at the target site was ~20%, and the corrected cells had higher ATP content, indicating the potential of mitoBE for treating mitochondrial genetic diseases (Fig. 4).
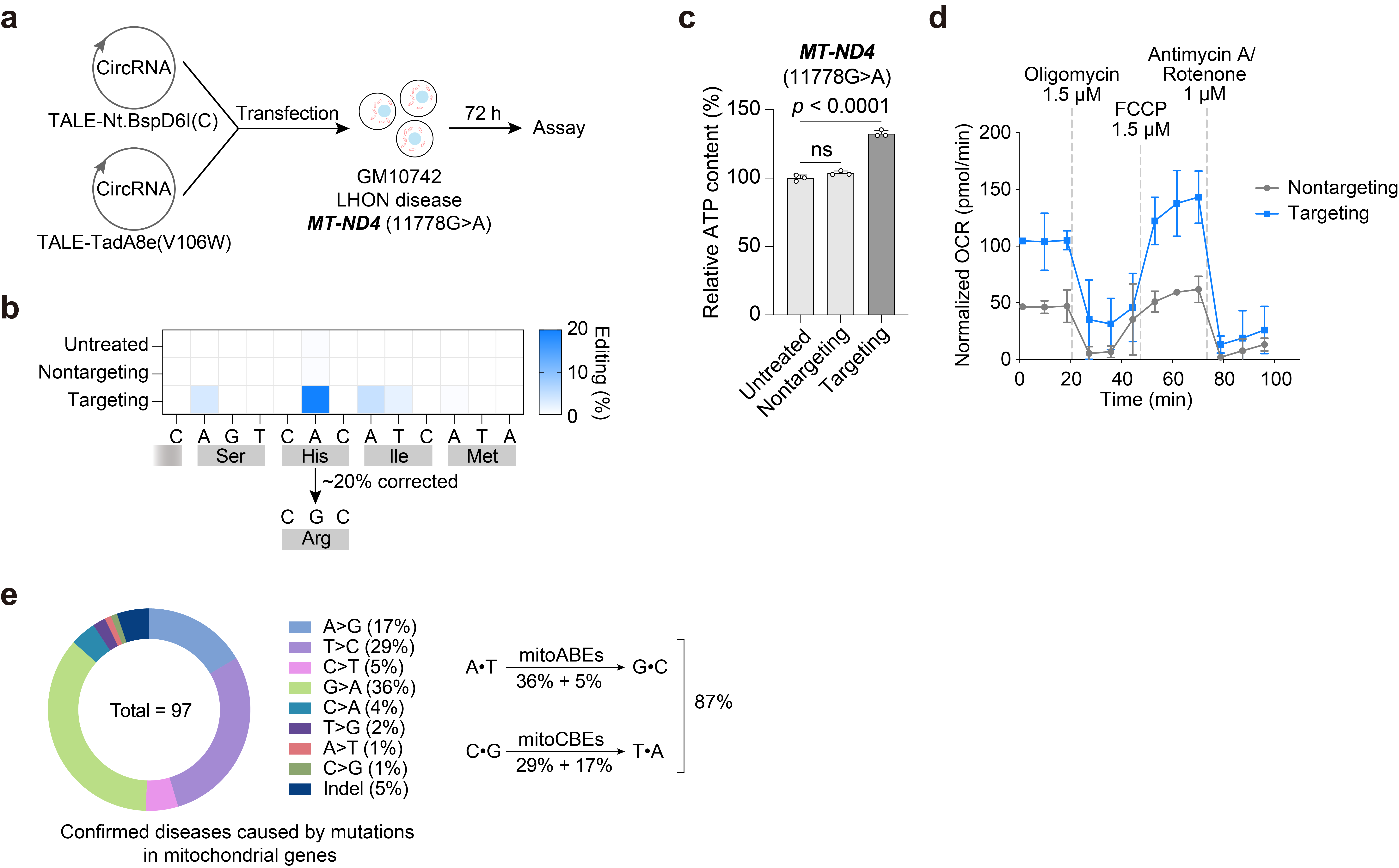
Fig.4 | Circular RNA-encoded mitoABEs successfully corrected the mutation in the LHON patient-derived disease cells. a, Overview of circRNA-encoded mitoABENt.BspD6I(C) transfected LHON disease cells GM10742, and genomic DNA was collected 3 days post-transfection. b, The editing efficiency of mitoABENt.BspD6I(C) corrected the 11778G>A mutation of LHON disease cell GM10742. c, The ATP levels of cells transfected with circRNA-encoded mitoABENt.BspD6I(C) targeted the 11778G>A mutation of LHON disease cell GM10742. Student’s T-test, p-value = 6.98E-05. d, OCR of the LHON disease cell line GM10742 treated with circRNA-encoded mitoABENt.BspD6I(C) targeted the 11778G>A mutation for 2 days. e. Types of mitochondrial diseases (MITOMAP) and the proportion of diseases that can theoretically be treated by mitoBEs. For b, the mean values from n = 3 biologically independent replicates are shown. For c and d, the data are presented as mean values ± s.d. of n = 3 independent biological replicates.
Using nickase to generate transient ssDNA deaminase substrates is an effective strategy for editing DNA. Theoretically, mitoBE can correct 84 out of 97 mitochondrial disease mutations, providing a promising new method for treating these illnesses (Fig. 4). What's more, our approach also works in the nucleus genome. The application of mitoBE to rectify errors in the mitochondrial and nuclear genome for treating diseases shows great potential.
Link:https://www.nature.com/articles/s41587-023-01791-y
Reference:
1 Vafai, S. B. & Mootha, V. K. Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374-383, doi:10.1038/nature11707 (2012).
2 Gammage, P. A., Moraes, C. T. & Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet 34, 101-110, doi:10.1016/j.tig.2017.11.001 (2018).
3 Bacman, S. R. et al. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med 24, 1696-1700, doi:10.1038/s41591-018-0166-8 (2018).
4 Gammage, P. A. et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med 24, 1691-1695, doi:10.1038/s41591-018-0165-9 (2018).
5 Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824-844, doi:10.1038/s41587-020-0561-9 (2020).
6 Deng, D. et al. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335, 720-723, doi:10.1126/science.1215670 (2012).
7 Mak, A. N., Bradley, P., Cernadas, R. A., Bogdanove, A. J. & Stoddard, B. L. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335, 716-719, doi:10.1126/science.1216211 (2012).
8 Yang, L. et al. Engineering and optimising deaminase fusions for genome editing. Nat Commun 7, 13330, doi:10.1038/ncomms13330 (2016).
9 Mok, B. Y. et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631-637, doi:10.1038/s41586-020-2477-4 (2020).
10 Cho, S. I. et al. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 185, 1764-1776 e1712, doi:10.1016/j.cell.2022.03.039 (2022).
11 Mi, L. et al. DddA homolog search and engineering expand sequence compatibility of mitochondrial base editing. Nat Commun 14, 874, doi:10.1038/s41467-023-36600-2 (2023).
12 Mok, B. Y. et al. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat Biotechnol 40, 1378-1387, doi:10.1038/s41587-022-01256-8 (2022).
13 Lei, Z. et al. Mitochondrial base editor induces substantial nuclear off-target mutations. Nature 606, 804-811, doi:10.1038/s41586-022-04836-5 (2022).
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


![When PSMA-targeted therapy is not enough: high-risk localized prostate cancer after repeated [177Lu]Lu-PSMA radioligand therapy](/cdn-cgi/image/metadata=copyright,fit=scale-down,format=auto,quality=95,width=256,height=256/https://public-storage.zapnito.com/Ku6h7Yyp4Q0LXqRRMICCHR2v4LcOsmxMrmDPtOYuI1c)



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in